|
1. Khái quát về các phương pháp phân loại vi sinh vật truyền thống:Trước đây công tác phân loại vi sinh vật vẫn dựa căn bản trên các đặc tính hình thái, sinh lý và hóa vi sinh vật: nhuộm, hình dạng tế bào khuẩn lạc, khả năng di động, nhu cầu dinh dưỡng, khả năng sinh acid trong môi trường cũng như sắc tố tạo thành v.v..Các đặc trưng này đôi khi cũng bộc lộ những hạn chế do các đặc tính được dùng cho nhóm vi sinh vật này (Enterobacteriaceae) nhưng lại không có ý nghĩa đối với nhóm khác (vi khuẩn gram âm - gram negative bacteria). Hạn chế của các phương pháp phân loại truyền thống dẫn đến nhiều trường hợp phải xác định lại tên phân loại của một số vi sinh vật. Từ trước đến nay, đơn vị cơ bản của định tên vi sinh vật là loài, bao gồm nhóm các cơ thể có mức độ tương đồng cao về các đặc điểm hình thái. Các phương pháp dựa trên các phản ứng sinh hóa: Từ những hạn chế của việc xác định các đặc tính hình thái dẫn đến nhiều nghiên cứu tập trung vào các phản ứng sinh hóa đặc trưng cho các vi sinh vật riêng biệt. Sự khác biệt của các phản ứng có ý nghĩa cho phân loại các vi sinh vật. - API20E KIT,nguyên tắc: dựa vào 20 phản ứng khác nhau. Nói chung hiện nay có nhiều nơi vẫn dùng kỹ thuật này nhưng nhìn chung kết quả cũng còn nhiều sai số do nhiều nguyên nhân khác nhau: cụ thể trong các trường hợp gene quyết định phản ứng sinh hóa nằm trong plasmid lại bị mất do nhiều nguyên nhân khác nhau như tuổi tế bào, lượng giống cấy hay thay đổi trong quá trình nuôi cấy dẫn đến sai khác và làm sai kết quả. - Phân biệt bằng thực khuẩn thể:Các vi khuẩn có độ mẫn cảm với thực khuẩn thể khác nhau. Có thực khuẩn thể xâm nhiễm làm tan tế bào ngay lập tức để sau đó thực khuẩn thể nhân lên thành các hạt trong tế bào chủ, trong khi đó với một số vi khuẩn thì thực khuẩn thể xâm nhiễm nhưng lại không làm tan tế bào vi khuẩn và chúng cùng tồn tại với tế bào vật chủ. Dựa vào sự khác biệt này mà người ta dùng các thực khuẩn thể khác nhau để phân biệt các đối tượng vi khuẩn nghiên cứu. Tuy nhiên, phương pháp này cũng bộc lộ một số nhược điểm khó giải quyết là các đặc tính mẫn cảm của vi khuẩn với thực khuẩn thể lại thay đổi do điều kiện ngoại cảnh hoặc là vi khuẩn lại có mức độ mẫn cảm khác nhau đối với các thực khuẩn thể khác nhau. Mặt khác nữa, thực khuẩn thể rất dễ thay đổi các đặc tính do đó cũng làm thay đổi cơ chế xâm nhiễm vào vi khuẩn chủ. - Phân biệt theo Typ huyết thanh:Đây là phương pháp được dùng khá lâu nhưng rất hiệu quả và hiện vẫn đang được sử dụng (ví dụ nhóm vi khuẩn Bt). Nguyên tắc là dựa vào nhóm quyết định kháng nguyên trên tế bào vi sinh vật (bề mặt tế bào tiên mao hoặc protein vỏ). Ưu thế của phương pháp này là các kháng huyết thanh được dùng để biệt hóa nhiều chi khác nhau, trong nhiều trường hợp đặc trưng cho loài. Nói chung đây là phương pháp khá ổn định nhưng hạn chế chủ yếu của phương pháp này ở chỗ: yêu cầu kỹ thuật sản xuất kháng huyết thanh và tiêu chuẩn hóa phản ứng kháng huyết thanh không đồng nhất tại các phòng thí nghiệm và tính ổn định giữa các lần lặp lại.
- Phân biệt bằng loại hoạt chất kháng khuẩn (Bacteriocin):Bacteriocin bản chất là peptid kháng khuẩn sinh ra bởi vi khuẩn để chống lại vi khuẩn khác. Như vậy, loại vi khuẩn tạo ra loại bacteriocin nào thì có khả năng kháng lại chính bacteriocin đó. Bởi vậy có nhiều loại vi khuẩn đã được phân loại dựa vào kiểu bacteriocin. Kết luận: Có nhiều phương pháp truyền thống được sử dụng trong nhiều nghiên cứu phân loại nhưng không có phương pháp nào tỏ ra vạn năng thích hợp cho mọi đối tượng vi sinh vật, tính chính xác chỉ có thể đạt được khi kết hợp nhiều phương pháp khác nhau.
2. Cách tiếp cận phân loại học với kỹ thuật sinh học phân tử:Ngày nay những tiến bộ trong sinh học phân tử đã mở ra khả năng ứng dụng hữu hiệu trong phân loại học và nghiên cứu đa dạng vi sinh vật. Nếu như các phương pháp truyền thống chỉ tập trung trên một số đối tượng vi sinh vật thì phương pháp sinh học phân tử có thể áp dụng trên mọi đối tượng vi sinh vật. Nói chung các phương pháp sinh học phân tử tập trung vào các kỹ thuật chủ yếu là: + Phân tích acid nucleic. + Phân tích protein. + Phân tích lipopolysaccharid. + Hóa phân loại học. Trong phần này chúng tôi tập trung giới thiệu một số kỹ thuật phân loại liên quan đến acid nucleic. Các phần sau chúng tôi lần lượt giới thiệu các phương pháp liên quan đến polysaccharid, lipoprotein và hóa phân loại. 2.1. Phân tích acid nucleic:Kỹ thuật phân tích acid nucleic liên quan chủ yếu đến phân tích kích thước, cấu trúc acid nucleic, mối tương quan về trình tự acid nucleic thông qua giải trình tự ADN và lai ADN. a. Phân tích ADN plasmid:Phương pháp phân tích ở đây bao gồm tách plasmid, so sánh các loại plasmid về kích thước sau đó tinh sạch plasmid và xử lý bằng enzym cắt hạn chế, sau đó điện di trên gel agarose và so sánh sự đa hình của các mảnh cắt để phân biệt các chủng vi sinh vật với nhau.
Plasmid là nhân tố di truyền ngoài nhân và sao chép độc lập với nhiễm sắc thể. Cấu trúc plasmid là sợi đôi ADN khép kín theo vòng. Tuy nhiên, có nhiều trường hợp ngoại lệ là sợi kép ADN mạch thẳng (đối với vi khuẩn Borrelia và Streptomyces). Plasmid được tìm thấy hầu hết ở các vi khuẩn và một số ít các vi sinh vật nhân thực bậc thấp như nấm men. + Tách plasmid:Thông thường lượng plasmid có trong tế bào chiếm < 5% tổng số acid nucleic của tế bào. Như vậy, ta phải thực hiện phép tách plasmid ra khỏi ADN của nhiễm sắc thể. Nói chung có nhiều phương pháp tách plasmid từ vi khuẩn nhưng nguyên tắc chung là phá tế bào vi khuẩn dùng enzym, siêu âm hay trong dung dịch kiềm có chất tẩy rửa là SDS hoặc TritonX-100, sau đó là việc loại ADN của nhiễm sắc thể và protein. Thông thường dùng phương pháp kết tủa với axetat. Lúc này phần lớn các thành phần ADN nhiễm sắc thể và protein của tế bào đã bị kết tủa bị loại bằng li tâm và plasmid nằm trong dịch trên tủa được tách khi kết tủa với ethanol hay isopropanol. Đây là phương pháp có hiệu quả để tách plasmid, trên thực tế hiệu quả tách các plasmid có kích thước nhỏ cao hơn nhiều so với các plasmid có kích thước lớn (>100 Kb). Với các plasmid có kích thước lớn cần các thao tác nhẹ nhàng để tránh đứt gãy do các nguyên nhân cơ học. Với cách tách plasmid như trên thì sản phẩm thu được có lẫn các đoạn ADN có kích thước khoảng 500 bp và nhiễm ARN. Để tránh nhiễm ARN thì cần xử lý với RNase (ribonuclease A). Tuy nhiên, có thể tách theo các phương pháp như: - Tách bằng Caeseium chloride. - Tách bằng KIT QIAGENE. - Tách bằng kỹ thuật khác.
+ Điện di plasmid trên gel agarose:
Sau khi thu được plasmid thì phải tiến hành kiểm tra trước khi phân tích kết quả điện di trên gel agarose để biết phổ plasmid và kích thước của chúng. Khi tiến hành điện di thì nồng độ gel khoảng 0.7- 1% với một trong các đệm sau: TAE (40 mM Tris acetat 1mM EDTA, pH 8.0), TBE (89 mM Tris borat 89 mM boric acid, 2 mM EDTA, pH 8.0) và TPE (80 mM Tris- phosphats 2 mM EDTA, pH 8.0). Sau khi điện di, gel được nhuộm với ethidium bromide và phát hiện dưới tia UV (310 nm). Nồng độ ethidium bromide khoảng 5 mg/l và thời gian nhuộm là 10 phút. Sau đó được rửa một lần nhanh bằng nước loại ion trước khi được nhìn dưới UV và chụp ảnh làm tài liệu. Một số vấn đề cần lưu ý khi phân tích plasmid: trên gel thông thường plasmid được thấy ở 3 dạng: dạng siêu xoắn, dạng đứt gãy và dạng mạch thẳng khi bị cắt bởi endonuclease. Dạng di chuyển tốc độ cao hơn là siêu xoắn (Hình 1.1).
Hình 1.1. Di chuyển của plasmid mạch thẳng (L) và siêu xoắn (OC) khi điện di Đôi khi việc tách plasmid cũng phức tạp đặc biệt trong những trường hợp plasmid có kích thước nhỏ có lẫn các mảnh ADN của nhiễm sắc thể. Trong mọi trường hợp đều có nhiễm các RNA và chúng di động nhanh nhất trừ các trường hợp plasmid như vậy không được nhầm lẫn giữa plasmid siêu xoắn và dạng chính plasmid đứt gãy và plasmid khác. Một chú ý khác là tránh nhầm lẫn khi so sánh kích thước giữa các plasmid siêu xoắn và dạng duỗi xoắn hay mạch thẳng. Thực tế là chỉ có thể so sánh các plasmid siêu soắn với nhau về kích thước. Khi tiến hành phân biệt giữa các chủng vi khuẩn thì đương nhiên là chỉ có thể thực hiện so sánh giữa các chủng cùng có plasmid với nhau. Cũng nên chú ý là không phải các chủng đều giống nhau về đặc tính miễn dịch nếu có chung kết quả phân tích plasmid như nhau. Phép phân tích sẽ có hiệu quả trong các mẫu có nhiều loại plasmid, tuy nhiên cũng phải tính đến việc các plasmid cũng có thể bị mất trong quá trình bảo quản. + Kỹ thuật dấu vân tay (finger printing) phân tích plasmid với enzym cắt hạn chế:Cho đến nay, người ta đã tìm thấy hơn 400 enzym cắt hạn chế. Mục đích của kỹ thuật này bổ sung cho kỹ thuật so sánh phổ kích thước plasmid ở trên. Tức là người ta có thể phân biệt được sự khác nhau của các plasmid có cùng kích thước. Như vậy, khi xử lý plasmid với một hay kết hợp một số enzym cắt hạn chế thì kết quả sẽ thu được là các đoạn ADN được cắt có kích thước khác nhau. Kết quả này có độ lặp lại cao, do đó dễ sử dụng khi so sánh các mẫu khác nhau. Hiện nay, có nhiều enzym cắt hạn chế được sản xuất và tiêu thụ trên thị trường, các enzym này có điểm nhận biết thay đổi từ 4-6 cặp nucleotid. Thông thường xử lý enzym trong 1 giờ sau đó mẫu được phát hiện trên gel agarose hoặc polyacrylamid Tuỳ thuộc vào kích thước của các mảnh cắt mà sử dụng agarose có nồng độ khác nhau: Bảng 1.1. Nồng độ agarose và kích thước mẫu ADN tương ứng.
Theo kinh nghiệm của nhiều tác giả (Grimont-1991) phổ các băng ADN có ý nghĩa cho so sánh không nên dưới 10 băng và nên có cả băng kích thước nhỏ và kích thước lớn. Tuy nhiên, các băng lớn không nên vượt quá 10 do băng có kích thước lớn như vậy khó di chuyển trên gel. Trong các trường hợp so sánh thì nên dùng thang chuẩn ADN để so sánh kích thước và phép phân tích dấu vân tay cho các lần phân tích khác nhau. Như đã trình bày ở trên, nếu như phân tích các chủng vi sinh vật dựa vào kích thước plasmid có ý nghĩa đối với các đối tượng có nhiều plasmid thì phương pháp xử lý enzym cắt hạn chế lại có ý nghĩa lớn trong các trường hợp nghiên cứu dịch tễ học trên các chủng có cùng phổ plasmid hay plasmid có kích thước giống nhau. Người ta đã ứng dụng kỹ thuật này trong nghiên cứu chủng E.coli (0157: H7) gây bệnh từ thực phẩm (Hamburger). Các plasmid từ các chủng khác nhau thì có phổ khác nhau về các đặc điểm cắt bởi enzym cắt hạn chế. Kết quả tạo ra các mảnh ADN đặc trưng cho cá thể làm cơ sở cho phép so sánh. Tuy nhiên khó có thể so sánh kết quả thu được từ các phòng thí nghiệm khác nhau, sở dĩ như vậy là do không dùng cùng một loại enzym cắt hạn chế. Một lý do nữa cũng cần được đề cập đến là sự xuất hiện các đột biến ngẫu nhiên tại vị trí enzym cắt, kết quả này tạo ra sự khác biệt về phổ thu được từ các mảnh cắt. b. Phân tích ADN nhiễm sắc thể:Phương pháp phân tích ở đây bao gồm việc tách ADN của nhiễm sắc thể và cắt bằng enzym cắt hạn chế để phân biệt giữa các vi sinh vật với nhau. Một chú ý khi tách ADN nhiễm sắc thể phải thao tác nhẹ nhàng để tránh đứt gãy do nguyên nhân cơ học. Nói chung các mảnh cắt nên có kích thước nhỏ hơn hoặc bằng 50 kb. Việc tách các mảnh cắt đó được thực hiện dựa vào kỹ thuật điện di trong trường xung điện (PFGE = Pulsed Field Gel Electrophoresis ).style="text-align: justify; margin-top: 6.0pt"> Như vậy kỹ thuật dấu vân tay không chỉ áp dụng trong trường hợp trên tế bào mang plasmid mà kỹ thuật này còn được sử dụng với nhiễm sắc thể cho mọi trường hợp.rường hợp.ường hợp.ường hợp. Liên quan đến vấn đề này phải kể đến kỹ thuật BRENDA: phân tích kết quả xử lý enzym cắt hạn chế với vi sinh vật. Ở đây cách chọn loại enzym cắt hạn chế vô cùng quan trọng, theo cách chọn này có thể tạo ra quá nhiều mảnh cắt nên không thể phân biệt được các băng riêng rẽ trong khi đó đối với các trường hợp khác lại tạo ra quá ít mảnh cắt có kích thước lớn khó tách theo các kỹ thuật điện di hiện có. Các vi sinh vật khác nhau có tỷ lệ GC khác nhau (dao động từ 25-75%) các mảnh cắt thu được sau khi xử lý enzym cắt hạn chế rất khác nhau. Theo Nei M. và Li W. H. (1979) thì số mảnh cắt có thể thu được tính trên lý thuyết là: a = (g/2)r1 x {1-(g/2)}r2Trong đó: g - tỷ lệ GC của ADN r1 - số cặp GC r2 - số cặp AT trong vị trí cắt của enzym giới hạn sử dụng a - số vị trí cắt khi sử dụng enzym cắt hạn chế. Mặt khác, người ta cũng căn cứ vào kết quả nghiên cứu các vị trí cắt của bộ gene vi sinh vật làm cơ sở cho cách chọn enzym cắt. Thông thường cho mỗi phép phân tích người ta ghi được số các băng cắt bởi enzym và tính toán kích thước các mảnh tạo thành làm cơ sở cho các phép phân tích về sau. Tuy nhiên, một trong những nguyên nhân hạn chế cho phép phân tích trên là các phương pháp tách ADN thông thường đều dẫn đến thay đổi kích thước do đứt gãy ADN nhiễm sắc thể, mặt khác sự có mặt của plasmid lẫn cũng tạo ra sự khác biệt giả trong kết quả phân tích, để khắc phục nhược điểm trên người ta phải thực hiện phép phân tích ADN nhiễm sắc thể nguyên vẹn để phát hiện các nguồn ADN tạp nhiễm lẫn vào kết quả phân tích. + Kỹ thuật điện di trong trường xung điện (điện di trong trường điện thay đổi, PFGE)- Nguyên tắc: Trước tiên các mẫu đưa vào phân tích phải được xử lý trước bằng loại enzym cắt hạn chế mà có rất ít điểm cắt. Kết quả phải tạo ra được các mảnh có kích thước lớn (50 kb – 12 Mb), với kích thước này không thể điện di theo phương pháp thông thường mà chỉ có thể tách ra bằng kỹ thuật PFGE (ật PFGE (ật PFGE (uật PFGE (Pulsed-Field Gel Electrophoresis ). Kỹ thuật PFGE lần đầu tiên được Schwartz va Cantor (1984) giới thiệu. Tuy nhiên sau đó đã có nhiều tác giả mô tả lại kỹ thuật này, tuy có sai khác ít nhiều nhưng cơ sở lý thuyết của các phương pháp này là như nhau: Mục đích của kỹ thuật này là tăng khả năng di động của các mảnh ADN có kích thước lớn trong điện trường, do đó các thành phần gel và đệm hầu như không thay đổi theo các phương pháp điện di thông thường. Tuy nhiên theo phương pháp thông thường thì sự điện di liên quan đến sự di động của các mảnh ADN có kích thước khác nhau trên gel agarose trong một trường điện không đổi. Kết quả ở đây là phân tử lớn khó di động hơn phân tử nhỏ qua mắt xích agarose, tạo ra sự tách biệt trong trường điện di. Trong trường hợp PFGE lực làm thay đổi khả năng di động lại là trường điện tích thay đổi liên tục dẫn đến các mảnh ADN có kích thước khác nhau thay đổi hướng di động. Như vậy kết quả là các mảnh có kích thước khác nhau sẽ có khả năng di động khác nhau. Kích thước càng lớn thì mức độ di động càng chậm. Sự di động khác nhau của ADN chủ yếu phụ thuộc vào kích thước chứ không phải do gel agarose như ở phương pháp điện di thông thường, hình 1.2.
Hình 1.2. Sử dụng kỹ thuật PFGE phân tích nhiễm sắc thể sau khi xử lý với Sma I với 7 chủng Haemophilus ìnluenzae. Tiếp theo các mô tả của Schwartz va Cantor, Smith và Codemine (1990) đã đề cập đến sự di chuyển của các mảnh ADN khác nhau là hàm số tuyến tính với kích thước của chúng. Trên cơ sở đó có thể điều chỉnh các xung điện và điện trường. Tuy nhiên các nhân tố khác cũng có mối tương tác lẫn nhau và ảnh hưởng đến khả năng di động của ADN như: nhiệt độ, điện thế, nồng độ agarose cũng như nồng độ ion trong đệm. - Chuẩn bị ADN cho PFGE: Chuẩn bị ADN từ nhiễm sắc thể không giống như các phương pháp chuẩn bị ADN plasmid. Một vấn đề thường xảy ra là sự đứt gãy ngẫu nhiên ADN dẫn đến sai khác trong kết quả phân tích. Để hạn chế điều này người ta phải thực hiện kỹ thuật tách ADN NST khỏi tế bào mẫu agarose có nhiệt độ nóng chảy thấp – LMT (Schwartz va Cantor – 1984 và Smith, Klco 1988). Theo phương pháp này hỗn dịch tế bào (dịch nuôi cấy hoặc dịch huyền phù) được trộn với LMT agarose tại 37oC. Hỗn dịch đầu tiên được xử lý với enzym và chất tẩy rửa để tách ADN NST khỏi thành, màng tế bào, RNA và protein. Sau đó là xử lý với EDTA, bất hoạt nuclease tế bào với proteinase K và N-lauroylsarcosine để loại các thành phần khác của tế bào. Kết quả là phần LMT agarose có chứa ADN NST được dùng làm mẫu chạy gel 0.5-1.0% (nên làm tan ở 65oC) và đưa lên mẫu chạy gel PFGE, tài liệu của Smith (1988) mô tả chi tiết cách chuẩn bị mẫu từ các tế bào có và không có thành tế bào: vi khuẩn, nấm men, nấm sợi, tế bào thực vật và động vật. Nói chung với các trường hợp có thành tế bào thì cần đến enzym phá thành tế bào. Lượng ADN NST thường dao động trong khoảng 0.5-20 µg ADN là thích hợp. Nếu quá nhiều ADN thì không thể phân tích được, còn quá ít thì cũng không thể phát hiện được các băng ADN trong kết quả phân tích. Mẫu ADN dùng cho kỹ thuật PFGE có thể được giữ 1 năm trong lạnh có chứa 0.5M EDTA. - Sau khi thu được ADN NST trong mẫu LMT agarose thì tiến hành xử lý với enzym giới hạn loại có ít điểm cắt. Tuy nhiên mẫu đầu tiên phải được xử lý với PMSF để bất hoạt protein K sau đó PMSF lại phải loại bỏ sau một số lần rửa với chất tẩy rửa và EDTA. Sau đó chỉ cần phân nhỏ mẫu trong agarose được xử lý với đệm và enzym cắt hạn chế qua đêm trong tube và sau đó toàn bộ mẫu này được sử dụng để tách trong trường xung điện. Bảng 1.2 dưới đây liệt kê các enzym cắt hạn chế được dùng cho phân tích PFGE. Bảng 1.2. Một số enzym cắt hạn chế được dùng cho kỹ thuật PFGE.
- Ứng dụng của kỹ thuật phân tích PFGE:
Kỹ thuật PFGE đã được sử dụng cho nghiên cứu trên nhiều đối tượng khác nhau (xem bảng 1.3). Bảng 1.3. Minh hoạ các chủng vi sinh vật được phân tích dựa trên kết quả PFGE thu được từ các đoạn nhiễm sắc thể.
Mỗi một đối tượng có đặc trưng riêng về kết quả phân tích PFGE và được dùng cho nghiên cứu so sánh với nhau về sự tương đồng. Đối với nhiều đối tượng có nhiễm sắc thể thì không nhất thiết phải thực hiện đối với mọi nhiễm sắc thể mà chỉ cần trên một số nhiễm sắc thể mà thôi. Ví dụ trong trường hợp của C.albicans Monod (1990) chỉ cần thực hiện phép phân tích với một trong 4 tổ hợp của một số nhiễm sắc thể là đủ. Cho dù theo các cách chuẩn bị nhiễm sắc thể khác nhau thì kết quả của phép phân tích PFGE đều giống nhau. Phép phân tích PFGE được ứng dụng cho các nghiên cứu ở mức độ loài với loài. Một số điểm cần lưu ý khi sử dụng kỹ thuật PFGE:+ Kỹ thuật này đòi hỏi phải có trang thiết bị chuyên dụng, kỹ thuật cũng như kinh nghiệm vì việc tách nhiễm sắc thể đôi khi gặp khó khăn trên một số đối tượng vi sinh vật. + Kỹ thuật PFGE không đủ nhạy để phát hiện những sai khác nhỏ giữa các mẫu, đặc biệt khi thực hiện với một enzym thì kết quả có thể giống nhau nhưng không chắc chắn là hai mẫu giống nhau hoàn toàn. Điều đó có nghĩa là kỹ thuật này nên dùng để xác định sự khác nhau còn việc xác định sự giống nhau thì không đủ tin cậy. Tuy nhiên ngay cả khi xác định sự khác nhau đôi khi các mẫu có plasmid nhỏ hay thực khuẩn thể cũng cần xét đến vì chính nguồn ADN này cũng tạo ra những sai khác giả. + Sự khác biệt giữa các mẫu có thể phát hiện được khi dùng các enzym cắt hạn chế khác nhau. Tóm tắt:Phương pháp phân tích plasmid là phương pháp được dùng phổ biến trong các phòng thí nghiệm chuẩn đoán vi sinh vật. Việc kết hợp giữa phân tích plasmid với sử dụng enzym cắt hạn chế sẽ tạo được sự phân tích chính xác với các thông tin đáng tin cậy đối với các mẫu phân tích. Tuy nhiên, khi phân tích các mẫu dựa trên plasmid thì cần lưu ý là các plasmid có thể bị mất qua các thế hệ phân chia, dẫn đến sai lệch các kết quả nghiên cứu. Khi thực hiện phép phân tích PFGE với các plasmid lớn sẽ khắc phục được hạn chế của các phép phân tích hiện tại với plasmid nhỏ như trên. Phương pháp PFGE hiện đang được dùng rộng rãi tại các phòng thí nghiệm vi sinh vật, áp dụng trên các đối tượng dễ nuôi cấy có thể cho các kết quả tốt hơn nhưng hạn chế về giá thành thiết bị cũng như yêu cầu cao về kỹ thuật và kinh nghiệm. Mặt khác khi sử dụng các enzym cắt hạn chế hiếm có thể sẽ khó phát hiện được mức độ sai khác. Như vậy sự kết hợp giữa các phép phân tích plasmid, PFGE và lai ADN sẽ cho kết quả tin cậy hơn. 2.2. Lai ADNNguyên tắc được tiến hành như sau: ADN tổng số được tách ra và xử lý với enzym cắt hạn chế (có trường hợp không cần xử lý với enzym cắt hạn chế) sau đó điện di trên gel agarose và chuyển lên màng lai. Mẫu dò (probe) được chuẩn bị và lai với màng ADN ở trên. Kết quả phép lai cho biết sự khác biệt giữa các mẫu ADN khác nhau. Phép lai được tiến hành ở đây dựa vào cấu trúc sợi đôi ADN từ hai sợi đơn với các cầu liên kết hydrogene theo nguyên tắc bổ sung. Bắt đầu là đứt gãy các liên kết hydrogene do hoá chất (kiềm) hay biến tính nhiệt, lúc này hai sợi đơn ADN tách nhau được gọi là trạng thái biến tính ADN (denature). Nếu tại thời điểm này người ta cho vào một ADN đánh dấu biến tính (mẫu dò) sau đó tạo điều kiện hồi biến xuất hiện (renature), lúc đó sẽ cho phép các sợi đơn của mẫu dò bắt cặp với các sợi đơn của mẫu ADN ban đầu (target ADN). Mức độ lai sẽ phụ thuộc vào tính đặc hiệu (sự tương đồng) giữa mẫu dò và mẫu gốc cũng như điều kiện cho phép lai thực hiện (nhiệt độ, nồng độ muối, pH, tỷ lện mẫu dò, kích thước mẫu dò). Như vậy, việc chọn điều kiện chính xác cho phép lai có ý nghĩa rất quan trọng cho tính đặc hiệu của kết quả phép lai. Sau khi lai, bước tiếp theo là rửa bằng đệm thích hợp để loại mẫu dò dư và cuối cùng là phép đo bức xạ đánh dấu (tuỳ theo phương pháp đánh dấu phóng xạ hay hoá chất phát xạ huỳnh quang) để đánh giá mức độ tương đồng của phép lai. Nói tóm lại một số nhân tố tham gia vào kết quả của phép lai là: mẫu dò ADN, phương pháp đánh dấu ADN, mẫu ADN lai và điều kiện tiến hành và phương pháp đánh giá kết quả phép lai. + Chọn mẫu dò:Việc chọn mẫu dò có ý nghĩa quan trọng cho phép lai. Nhìn chung các nhà phân loại học vi sinh vật thường không trực tiếp thiết kế mẫu dò. Về mặt này chúng tôi đưa ra một số nội dung chủ yếu về tiêu chí chọn mẫu dò theo Stahl và Amann (1991) như sau: Mẫu dò sẽ lai với ADN đích (target) và không lai với các nguồn ADN khác có mặt trong mẫu lai. Khi phân tích các mẫu vi sinh vật với nhau thì mẫu dò nên là đoạn nucleotid đặc trưng cho các mẫu phân tích để phân biệt với các sinh vật khác. Tuy việc chọn mẫu dò cũng mang tính kinh nghiệm, mẫu dò đôi khi không cần phải là toàn bộ gene mà chỉ là một đoạn gene mã hoá cho một đặc tính đặc trưng cho đối tượng nghiên cứu (có thể là yếu tố kháng nguyên bề mặt, độc tố hay một gene chức năng nào đó trên plasmid). Với cách chọn mẫu dò có kích thước nhỏ (14-40 bp), có thuận lợi là các mẫu dò được tổng hợp nhân tạo và phép lai thực hiện nhanh (dưới 30 phút) trong khi đó với các mẫu dò lớn thời gian kéo dài hơn (khoảng 16 giờ). Hạn chế lớn nhất đối với các mẫu dò nhỏ là khó khăn khi đánh dấu và dẫn đến giảm tính đặc hiệu của phép lai. Một cách thường được sử dụng là thiết kế các mẫu dò từ các chuỗi ARN thông tin 16S. Cách chọn có thể căn cứ vào đoạn ADN có mặt trong các đại diện mức độ dưới loài, trong loài hay thuộc chi nghiên cứu. + Các phương pháp đánh dấu:Các kỹ thuật đánh dấu ADN được xem là lĩnh vực phát triển mạnh nhất trong sinh học phân tử. Nói chung kỹ thuật đánh dấu được chia thành kỹ thuật đánh dấu trực tiếp và gián tiếp. Trong phương pháp trực tiếp thì phần đánh dấu để phát hiện được gắn vào acid nucleic. Đối với phương pháp gián tiếp thì có một phần chức năng được gắn vào acid nucleic và phần này lại được phát hiện gián tiếp dựa vào protein bám đặc hiệu được phát hiện bằng kỹ thuật miễn dịch. Sau đây là chi tiết các phương pháp đánh dấu. · Đánh dấu trực tiếp:- Bảng dưới đây đưa ra các loại cơ chất dùng cho phương pháp đánh dấu trực tiếp dựa vào việc nhân ADN được đánh dấu lên sẽ tăng độ nhạy so với phương pháp đánh dấu chỉ được thực hiện với các mồi đơn lẻ. Đối với các phương pháp đánh dấu trực tiếp thì các nhóm tạo tín hiệu được gắn trực tiếp bằng liên kết hoá trị với nucleic của mẫu dò. - Đánh dấu trực tiếp bao gồm 2 bước chính: tạo tín hiệu dựa vào liên kết hoá trị của nhóm tín hiệu với mẫu dò và thực hiện phép lai giữa mẫu nghiên cứu và mẫu dò. Bảng 1.4. Một số cơ chất dùng cho đánh dấu trực tiếp.
Hình 1.3. Minh hoạ phương pháp đánh dấu trực tiếp (A) và gián tiếp (B)
· Đánh dấu gián tiếp:
Đánh dấu gián tiếp được thực hiện theo 3 bước: thực hiện phép lai giữa mẫu dò và mẫu ADN đích, phản ứng giữa nhóm chức năng và protein bám đặc hiệu với nhóm chức tín hiệu thông báo (reporter) và cuối cùng là tạo ra tín hiệu từ nhóm chức tín hiệu.
Bảng 1.5. Liệt kê một số hệ thống đánh dấu gián tiếp.
Mặc dù có nhiều hệ thống đánh dấu và phát hiện tín hiệu nhưng kinh nghiệm cho thấy hệ thống biotin và digoxidenin cho độ nhạy cao hơn cả. Hai hệ thống này có thể phát hiện tới mức dưới picrogam acid nucleic. Trong trường hợp với biotin thì đôi khi có mức độ nhiễu nền cao do biotin có mặt trong các sinh phẩm, còn đối với digoxigenein thì có thể không gặp khó khăn này. + Đánh dấu phóng xạ và ghi phóng xạ:Phương pháp đánh dấu phóng xạ là phương pháp được dùng phổ biến trong các phòng thí nghiệm lai ADN với các thành phần đánh dấu là 32P và 35S. Kết quả của phép lai giữa mẫu dò và ADN đích được phát hiện trên X-phim hoặc nhấp nháy lỏng. Ưu điểm nổi bật của phương pháp đánh dấu phóng xạ là độ nhạy có thể đạt tới dưới mức picrogam ADN đích. Hạn chế của phương pháp này là không đạt được sự thích hợp cần thiết cho các thí nghiệm chẩn đoán liên quan tới xác định mẫu vi sinh vật, ví dụ thời gian bán huỷ mẫu dò ngắn, nguy hiểm cho người sử dụng và cần yêu cầu an toàn nghiêm ngặt cho phòng thí nghiệm và cá nhân sử dụng cũng như việc quản lý chất thải. Xuất phát từ thực tế trên mà càng ngày người ta càng quan tâm đến các phương pháp đánh dấu phi phóng xạ. Mục tiêu là đạt được một hệ thống đánh dấu có độ nhạy tương đương với phương pháp dùng chất phóng xạ. Bảng dưới đây liệt kê các yêu cầu lý tưởng cho phương pháp đánh dấu mẫu dò: 1, Dễ gắn với acid nucleic theo phương pháp đơn giản và có độ lặp lại cao. 2, Bền trong điều kiện lai và bảo quản. 3, Không bị ảnh hưởng và làm ảnh hưởng đến phản ứng lai. 4, Có thể thực hiện với điều kiện phản ứng lai trong dịch thể (liquid phase) hay có chất mang (solid phase). 5, Phương pháp phát hiện nhạy và đơn giản. 6, Dễ bị loại bỏ sau phản ứng lai. 7, Dễ phân biệt giữa mẫu lai và mẫu chưa lai trong cùng điều kiện. 8, Có thể ứng dụng với hệ thống tự động hoá. Tuy nhiên, nếu theo các yêu cầu lý tưởng trên thì chưa có hệ thống đánh dấu phi phóng xạ nào thoả mãn. Hiện nay, có nhiều hệ thống đánh dấu phi phóng xạ đạt độ nhạy cao và đó là lý do các phương pháp này đang được ứng dụng rộng rãi đặc biệt là phương pháp dựa trên các enzym như Alkaline phosphatase, Horseradisk peroxidase. Tuy nhiên phương pháp sử dụng các chất phóng xạ hiện tại vẫn đang được dùng cho một số phòng thí nghiệm đặc biệt. · Chiến lược đánh dấu với chất phi phóng xạ.Có 3 phương pháp đánh dấu phi phóng xạ: 1, Dùng cơ chất hoá phát xạ và sinh phát xạ, trong trường hợp này thì cả cơ chất hoá và sinh phát quang đều tạo ra các bức xạ ánh sáng và sau đó là phương pháp phát hiện bức xạ này. * Với nguồn cơ chất hoá phát quang có loại như AMPPD (Bronstein, Edward & Voyta, 1989) có thể được dùng trực tiếp như là cơ chất cho enzym Alkaline phosphatase trong phép lai ADN đạt độ nhạy cao trong thời gian ngắn (Bronstein et al., 1990). * Phương pháp phát xạ sử dụng enzym Alkaline phosphatase trên cơ sở giải phóng D-luciferin từ cơ chất, ví dụ D-luciferin-O-phosphate (Miska và Geiger., 1987). Hai phương pháp này tương đối nhạy và đang được sử dụng rộng rãi. 2, Phương pháp phát hiện dựa vào thay đổi màu:Phương pháp đo màu có thể thực hiện trên môi trường dịch thể hay chất mang và khá nhạy so với phương pháp phát quang. Người ta đã tạo ra các cơ chất sinh màu khi có mặt enzym (chẳng hạn như Alkaline phosphatase). Lợi thế của phương pháp này là việc phát hiện đơn giản do kết quả là sự thay đổi màu dễ phát hiện và ứng dụng trong các phòng thí nghiệm vi sinh vật. Một phương pháp có thể làm tăng độ nhạy của phản ứng màu bằng cách thêm một enzym kích hoạt phản ứng màu vào hệ thống. Một ví dụ cho điều này là vai trò loại nhóm phosphat của phân tử NADP thành NAD do Alkaline phosphatase trong hệ thống phát hiện mẫu dò nucleic. Trong hệ thống này thì NAD có vai trò kích hoạt chu trình oxy hoá mà có sự tham gia của alcohol dehydrogenease và diaphorase. Trong mỗi một chu trình thì NAD sẽ bị khử thành NADPH + H+. Phản ứng oxy hoá này lại kèm theo phản ứng oxy hoá mà NADPH + H+ lại bị oxy hoá thành NAD, mọi phản ứng xảy ra gắn liền với việc khử ρ-indonitro-tetrazolium màu tím thành formazan. Sản phẩm cuối cùng formazan được định lượng bằng phép so màu (Self. 1985). 3, Cơ chất huỳnh quang và phát xạ huỳnh quang theo thời gian.Sử dụng cơ chất phát xạ huỳnh quang là phương pháp khá nhạy và có thể phát hiện được tới một phân tử chất phát xạ (fluorescein). Nói chung với các mẫu sinh phẩm thường có mức độ nhiễu tín hiệu nền cao. Thực tế này có thể được cải thiện với cơ chất fluorophore bền bị kích hoạt bởi sóng ánh sáng. Sự phát xạ sau đó được ghi lại khi các bức xạ nền đã bị giảm. Đối với phương pháp dùng Alkaline phosphatase thì việc phát hiện nhân tố gắn lanthanide theo thời gian đi kèm với hoạt tính enzym này gọi là phương pháp phát quang lanthanide khuyếch đại bởi enzym (EALL: Evangelista, Pollack & Templeton, 1991). Trong trường hợp này, cơ chất không có khả hình thành phức hệ gắn với lathanide phát xạ. Dưới tác dụng của Alkaline phosphatase thì cơ chất và lanthalide bị chuyển thành phức hệ hoạt động. Phương pháp này khá nhạy nhưng hạn chế lớn nhất của nó là cần thiết bị kích hoạt và đo bức xạ huỳnh quang. + Quá trình lai:Nói chung quá trình lai (Hybridization ) được tiến hành theo một trong 3 cách sau để định danh hay xác định vi sinh vật: Lai với vi sinh vật (in situ), lai trong dịch thể và lai với chất mang (solid support). a. Kỹ thuật lai với vi sinh vật (in situ), hình 1.4.
Hình 1.4. Minh hoạ kỹ thuật lai in situ với kỹ thuật FISH (fluorescence in situ hybridization) phát hiện vi khuẩn Helicobacter pylori. Kỹ thuật này được thực hiện để định vị chuỗi acid nucleic trong vi sinh vật sau khi đã được cố định trong các tiêu bản hiển vi. Tuy nhiên, do hầu hết các vi sinh vật có khả năng thấm (hay cho phép) các đoạn ngắn oligonucleotid đánh dấu đi vào tế bào sau khi cố định mẫu vì vậy khi sử dụng mẫu dò đánh dấu với chất nhuộm huỳnh quang đặc biệt có thể nhìn thấy được trực tiếp vi sinh vật dưới kính hiển vi huỳnh quang. Điều đáng chú ý là phản ứng lai phải tiến hành trong thiết bị được hàn kín nhằm hạn chế việc bay hơi của dịch lai. Việc bay hơi dịch lai này dẫn đến việc bám không đặc hiệu của chất nhuộm huỳnh quang với tế bào. Nhiệt độ lai phụ thuộc vào oligonucleotid mẫu dò. Sau phản ứng lai thì mẫu phải được xem ngay hoặc được bảo quản trong tối trong thời gian 6 tháng. Nếu mẫu dò đặc hiệu cho RNA thì độ nhạy tăng lên rất lớn vì có tới hàng ngàn bản copy của RNA trong mỗi tế bào (Stahl và Amman, 1991). Lai trong môi trường dịch thể:Phản ứng lai trong môi trường dịch thể xảy ra nhanh hơn nhiều so với môi trường có chất mang pha rắn (soild). Do cả phân tử ADN đích và mẫu dò đều có thể di động tự do trong môi trường do đó phản ứng đạt kết quả cao nhất. Nói chung với phép lai thực hiện trong dịch thể thì thời gian kết thúc nhanh hơn từ 5-10 lần so với trong điều kiện là chất mang (Bryan, et al., 1986). Tuy nhiên, cũng có thể bổ sung thêm chất mang làm tăng phản ứng lai. Cần thêm bước cuối cùng là tách phức hợp mẫu dò-sợi ADN đích. Hạn chế ở đây là mẫu trong dịch cho nên có thể tạo lên các nền kém đặc hiệu, do vậy cần các giải pháp làm hạn chế mức độ nhiễu của nền cho thích hợp.
Hình 1.5. Minh hoạ kỹ thuật lai trong môi trường dịch thể. Trên thực tế hầu hết các KIT thương phẩm dùng cho vi sinh vật đều tiến hành trong môi trường dịch thể. Vi sinh vật trong mẫu đầu tiên được phân giải trong điều kiện thích hợp để ADN đích lai với mẫu dò. Sau đó là bước lai và tách phức hệ mẫu dò và ADN đích dựa vào các thông số của phép lai theo kiểu kẹp díp cụ thể. Các phép lai theo kiểu kẹp díp (hình 1.9) cần có hai đoạn ADN có trình tự khác nhau; một đoạn để bám giữ ADN đích gắn vào chất mang và một đoạn là mẫu dò. Điều quan trọng là hai đoạn ADN này phải có trình tự khác nhau cho dù chúng đều gắn với hai vùng khác nhau trên đoạn ADN đích theo nguyên tắc bổ sung. Mẫu dò đánh dấu được bổ sung vào trong dung dịch có các đoạn ADN đích nghiên cứu. Như vậy theo hình vẽ phức hợp ADN mẫu dò đánh dấu để phát hiện gắn với ADN đích và phức hợp mẫu dò- ADN đích gắn vào chất mang đã được hình thành. Phức hợp này được tách ra khỏi dung dịch và khi có mặt cơ chất thích hợp để phát hiện được mẫu dò đánh dấu. Khi dùng hệ thống phát hiện dựa vào các hoá chất huỳnh quang hay tạo màu có thể dùng thiết bị phát hiện kết quả một cách tự động. + Kỹ thuật lai dựa vào màng:Tác giả đầu tiên giới thiệu kỹ thuật cố định ADN trên màng là Nygaard và Hall (1963, 1964). Sau đó chính Southern (1975) là người mô tả việc tách ADN khỏi gel sau khi điện di và chuyển chúng lên màng nitrocellulose. Các đoạn ADN đích được phát hiện bằng kỹ thuật lai và đây là bước tiến quan trọng của sinh học phân tử. Từ năm 1977 đến nay đã có nhiều cải tiến về phép lai cũng như bước chuyển ADN lên màng của các tác giả khác nhau như: Meinkoth và Wahl (1984). Người ta cũng đã sử dụng một số màng nynol, màng nitrocellulose hoạt hoá hay có độ bền cho các phép lai. Đối với công việc định loại vi sinh vật với kỹ thuật cố định trên màng cho phép lai ADN thì cần chấm mẫu (là ADN sạch, hay tế bào vi sinh vật hoặc sinh phẩm có vi sinh vật nghiên cứu) lên màng thích hợp. Mẫu được ly giải và ADN bị biến tính, sau đó ADN được cố định trên màng khi đưa vào tủ xấy tại 80oC trong 2 giờ hoặc đưa vào xử lý với tia UV trong trường hợp sử dụng màng lynon. Mẫu dò đánh dấu sau đó được đưa vào dung dịch. Bước tiền lai được thực hiện để hạn chế các mẫu bám vào nhau không đặc hiệu. Sau đó là bước lai, màng sau khi lai được rửa để loại các mẫu dò còn dư. Bước rửa tiếp theo để đánh giá mức độ bám đặc hiệu của phép lai. Sau đó các mẫu dò đánh dấu lai với vị trí ADN mẫu trên màng được phát hiện bằng các hệ thống phát hiện tương thích. Toàn bộ quá trình lai trên màng đã được Meinkoth và Wahl (1984) mô tả chi tiết. Khi phát hiện typ vi sinh vật, chúng ta phải sử dụng kỹ thuật Southern. Trong phương pháp này ADN của nhiễm sắc thể được tinh sạch và được xử lý với enzym cắt hạn chế thích hợp, sau đó mẫu lại được điện di trên gel agarose và tạo ra dấu vân (finger print) của nhiễm sắc thể. Sau khi điện di gel được đặt dưới màng nitrocellulose hay màng nylon nằm giữa một số lớp giấy. Lớp giấy lọc phía trên được đặt trên cùng phần gel và màng kẹp giữa giấy lọc như trong hình 1.6.
Hình 1.6. Mô tả phương pháp chuyển ADN lên màng cho kỹ thuật Southern Blot. Kết quả là đệm từ phía dưới được thấm một cách tự nhiên lên trên. Điều đó giúp cho việc chuyển các mảnh ADN từ gel lên màng và bám chặt vào màng với kích thước và phiên bản đúng như trên gel sau khi điện di (mô tả chi tiết theo Sambrook 1989). Phương pháp truyền thống này cũng được cải tiến khi dùng màng nylon để chuyển ADN trong điều kiện biến tính (Read và Mann 1985). Thời gian chuyển có thể vài giờ hoặc qua đêm. Việc cố định ADN trên màng được thực hiện sau đó bằng cách xử lý nhiệt hay tia UV như đã trình bày ở trên. Tuy nhiên, nhiều phương pháp cải tiến được thực hiện nhanh như chuyển bằng điện di (Bittner, Kupfere, Morris, 1980) hay sử dụng bơm chân không (Medveczky, 1987; Olszewsk, 1988). Trong nhiều trường hợp dùng điện để chuyển ADN từ gel agarose lên màng thì trước tiên gel phải được xử lý biến tính và được làm trung hoà trở lại. Gel được đặt giữa hai bản điện có các bản giấy thấm (hình 1.7).
Hình 1.7. Minh hoạ bước chuyển ADN lên màng. Sau đó nguồn điện được nối và lúc đó ADN sẽ được chuyển lên màng. Thời gian chuyển sẽ phụ thuộc vào kích thước đoạn ADN nhưng thường dao động trong khoảng 1-3 giờ. Phương pháp có thể thực hiện theo chiều thẳng đứng hay chiều nằm ngang. Hiện nay có một số thiết bị bán trên thị trường được dùng cho kỹ thuật này. Với thiết bị nằm thẳng đứng, thì toàn bộ được ngâm trong đệm và có thể tăng cường độ dòng điện (chú ý vì có thể làm tăng nhiệt độ), phương pháp này cần dùng nhiều đệm. Ngược lại theo phương pháp nằm ngang hay phương pháp semi-dry, ở đây gel và màng được kẹp giữa các bản giấy lọc đã tẩm ướt bằng đệm thích hợp. Trường hợp này cần rất ít đệm và cường độ dòng điện là 1mA/cm2 là thích hợp. Trong trường hợp chuyển ADN lên màng bằng áp lực do chân không, việc thiết lập hệ thống được trình bày mà ở đó gel được đặt trên màng và sử dụng lực hút chân không để đưa ADN lên màng. Dùng lực hút chân không đạt được hiệu quả chuyển ADN lên màng cao hơn phương pháp thấm tự nhiên. Hiệu quả tối đa cho chuyển gel với trường hợp geneom sau xử lý enzym cắt hạn chế có thể đạt được sau 1 giờ. + Phân tích RELPs với kỹ thuật lai ADN.Giới thiệu phương pháp: Như đã trình bày, sự phức tạp trong kỹ thuật RELP đã tạo ra những khó khăn cho việc xác định các tác nhân gây bệnh trong các nghiên cứu về dịch tễ học. Điều này còn khó khăn và phức tạp hơn trong khi so sánh các kết quả thu được trên các bản gel khác nhau hoặc tại các phòng thí nghiệm khác nhau. Nguyên nhân là do số lượng các băng ADN lớn tới mức không thể xác định kích thước của chúng hay các băng thu được không lặp lại các kết quả nghiên cứu. Mặc dù vậy, theo phương pháp này các phổ ADN phức tạp sau khi xử lý với enzym cắt hạn chế sẽ được làm biến tính và chuyển lên màng nitroxenluloza hay nylon. Màng sẽ được lai với mẫu dò thích hợp, kết quả phổ phép lai sẽ rõ và không quá phức tạp do nó chỉ hiển thị các mảnh ADN bắt cặp với mẫu dò. Với một số lượng không lớn các mảnh hiển thị thu được sau phép lai sẽ cho phép xác định được chính xác hơn về kích thước. Điều này làm cơ sở cho so sánh giữa các gel với nhau cũng như kết quả thu được từ các phòng thí nghiệm khác nhau. Khi sử dụng phương pháp này cần chú ý một số mẫu dò sau: 1, Mẫu dò có thể là đoạn RNA ribosom từ một loài sinh vật nào đó: ví dụ rRNA của E.coli có thể được các công ty thương phẩm cung cấp (Boeringer Mannheim), đây là mẫu dò khá thuận lợi và được đánh dấu phóng xạ hay với các cơ chất huỳnh quang. Ưu điểm chính của mẫu dò này ở chỗ phần RNA là đoạn khá bảo thủ do đó mẫu dò có thể dùng lai với các sản phẩm xử lý enzym cắt hạn chế của nhiều loài vi khuẩn khác nhau. Có một số loài khi lai với mẫu dò chỉ cho một kết quả giống nhau trong khi đó ở loài khác khi lai các chủng với mẫu dò thì lại thu được kết quả khác nhau. Đây là cơ sở cho phương pháp ribotyping. 2, Mẫu dò có thể là đoạn ADN ngẫu nhiên có chức năng không xác định (Tompkins et al., 1986). Mẫu dò này thường được dùng cho các loài có chứa chính các đoạn ADN này. Sử dụng các mẫu dò này đôi khi rất có ý nghĩa trong việc phân biệt các mẫu sau khi tiến hành phương pháp ribotyping (Saunders et al., 1990). 3. Một cách khác nữa cũng được dùng là sử dụng mẫu dò là một đoạn ADN tách dòng từ một gene đã biết. Ví dụ dùng đoạn gene mã hoá cho độc tố ngoại bào (exotoxinA) sử dụng để định typ Pseudomonas aeruginosa (Ogle et al., 1987). Mẫu dò dùng gene mã hoá cho độc tố Cholera được dùng để xác định các typ cho các chủng thuộc họ phảy khuẩn sinh độc tố (Yam, Li Lung & Ng, 1989). Việc sử dụng bất kỳ loại mẫu dò nào cũng tạo ra phổ đặc trưng của phép lai có ý nghĩa cho so sánh giữa các chủng với nhau. Hạn chế chính của kỹ thuật này ở chỗ kết quả thu nhận chỉ khu trú phần gene bắt cặp với mẫu dò mà không đặc trưng cho cả hệ gene. Bảng 1.6. Kết quả thu được khi thực hiện kỹ thuật RFLP với dùng các mẫu dò không phải là ribosom.
- Kỹ thuật ribotyping:Như đã trình bày ở trên mọi mẫu dò đều cho các kết quả có sức thuyết phục tuy nhiên kỹ thuật dùng mẫu dò là đoạn RNA của ribosom đã đưa ra một cách tiếp cận mới trong nghiên cứu dịch tễ phân tử với các vi khuẩn có sự khác biệt lớn trong khi đó các mẫu dò khác chỉ giới hạn với một loài hay chỉ có ý nghĩa cho các chủng trong cùng một loài. Kỹ thuật này lần đầu tiên được Grimont mô tả năm 1986 và nhanh chóng trở thành phương pháp hữu hiệu hiện nay cho nghiên cứu dịch tễ học vi sinh vật ở mức độ phân tử. Tính hợp lý cho việc sử dụng kỹ thuật này là ở chỗ gene mã hoá cho RNA ribosom có độ bảo thủ cao. Cũng có thể phát hiện thấy những thay đổi chút ít trong quá trình tiến hoá đối với các chuỗi ADN trong các vi khuẩn nghiên cứu. Gene RNA ribosom được tổ chức thành các operon mà các gene riêng rẽ mã cho các RNA kích thước 5S, 16S và 23S chúng được cách nhau bằng các đoạn ADN spacer không mã cho gene nào. Nếu dùng mẫu dò hỗn hợp giữa 16S và 23S thì kết quả phép lai sẽ hiển thị các mảnh tương ứng với phần của gene này trong khi dùng mẫu dò là đoạn của các gene đã được tách dòng có thể đưa đến kết quả hiển thị cả phần gene tương đồng và chuỗi spacer. Như vậy sự khác nhau của kết quả phụ thuộc vào loại mẫu dò sử dụng. - Yêu cầu kỹ thuật:Để thu được kết quả tối ưu cần có các yêu cầu kỹ thuật cụ thể cho từng bước thực hiện. Đầu tiên là phải tạo ra được phổ dấu vân tay thu được sau khi xử lý mẫu với enzym cắt hạn chế. Phổ vân tay tối ưu khi các mảnh cắt phải được tách rõ và phân bố đồng đều về kích thước trên gel. Số lượng các mảnh ADN thu được phụ thuộc vào mẫu ADN và loại enzym cắt hạn chế được sử dụng. Thông thường phải chọn một số mẫu xử lý thử trước với từng loại enzym (hay đồng thời xử lý với một số enzym). Có thể thấy rõ là các enzym có 5 nucleotid tại vị trí cắt sẽ tạo ra sản phẩm nhiều mảnh hơn các enzym có 6 nucleotid tại vị trí nhận biết. Thực tế cũng cho thấy khi dùng enzym cắt hạn chế (một hay đồng thời vài loại) để thu được phổ các đoạn cắt hợp lý cho việc phân tích ribotyping thì các kết quả này cũng rất khác nhau. Như vậy, điều quan trọng nên tiến hành trước các phép phân tích này, phải chọn được một số chủng đặc trưng và thử với một số enzym cắt hạn chế để chọn giải pháp cho nghiên cứu dịch tễ học với kỹ thuật này. Trong một số trường hợp kết quả phân tích khi chỉ tiến hành với 1 hay 2 enzym cắt hạn chế có thể không đưa ra được kết quả chính xác. Như vậy, khi có được kết quả hợp lý sau khi xử lý các mẫu ADN với enzym cắt hạn chế thì tiến hành các bước chuyển các đoạn ADN trên gel lên màng theo các phương pháp đã được mô tả ở trên. Phương pháp chuyển bằng chân không là hợp lý hơn cả vì kết quả cho việc chuyển các băng có kích thước gần nhau lại được tách ra sắc nét trên màng. Khi thực hiện phép lai nên chú ý nếu như dùng nguồn ARN ribosom của một chủng nào đó đánh dấu làm mẫu dò thì cần phải thay đổi điều kiện lai như nhiệt độ để phép lai được thực hiện tốt với các đoạn ADN từ các chủng vi sinh vật khác nhau. Có một số phương pháp đánh dấu mẫu dò là ADN ribosom. Phương pháp đánh dấu phóng xạ (Grimont, 1986), đây là phương pháp có ưu thế là chỉ cần vài bước đơn giản cho phép lai và rửa mẫu nhưng lại mang nhiều hạn chế như: thời gian bán huỷ ngắn, yêu cầu an toàn cho phòng thí nghiệm riêng biệt, nguy hiểm cho người dùng và gặp khó khăn với việc xử lý chất thải phóng xạ. Mới đây có một số phương pháp đánh dấu mẫu dò phi phóng xạ được sử dụng khá thành công. Pitcher (1987) đã mô tả phương pháp tổng hợp mẫu dò gắn với bilatin để phát hiện ARN ribosom của Providencia stuartii. Chất kích hoạt quang hoá đã được Koblavi và Grimont mô tả (1990). ARN ribosom cũng được đánh dấu bằng acetylaminofluorence (AAF) do Grimont (1989) mô tả. Công ty Eurogeneetik (Bỉ) đã cung cấp phức hợp AAF-rARN trên thị trường. Gần đây Gustafero và Persing (1992) đã đưa ra một phương pháp mới mà ở đây phức hợp horseradisk-peroxysase-polyethyleneimine với rARN và kích hoạt bằng quang hoá. Các mẫu dò được đánh dấu bằng các chất phi phóng xạ này có thể cho kết quả nhanh tương đương với trường hợp dùng các chất phóng xạ. Như vậy kết quả tiến hành phép lai các mẫu của các chủng khác nhau trong cùng gel chuyển lên có thể so sánh với nhau khi dùng kỹ thuật này. Trong trường hợp xác định chính xác kết quả các mảnh lai với mẫu dò có thể tiến hành so sánh các kết quả thu được từ các phòng thí nghiệm khác nhau. + Ứng dụng kỹ thuật ribotyping: Xác định typ vi khuẩn bằng kỹ thuật ribotyping có một số ưu điểm so với một số kỹ thuật định typ khác ở chỗ: Thực tế là các gene của ribosom khá bền do chúng nằm trên nhiễm sắc thể. Hầu hết các vi sinh vật đều chứa nhiều phiên bản của operon ribosom do đó khi lai với mẫu dò thích hợp thì kết quả là tạo ra một số mảnh lai có kích thước khác nhau. Hiện nay, nguồn rARN của E.coli đánh dấu là mẫu dò khá phổ biến đã được thương mại hoá.
Hình 1.8. Kết quả ribotyping với Helicobacter pylori. Do tính đa dạng và phức tạp của kỹ thuật định typ theo ribosom do đó trên thực tế khi nhìn kết quả bằng mắt thường khó có thể phát hiện được sự khác nhau hay giống nhau giữa các chủng trong cùng một loài khi tiến hành nghiên cứu dịch tễ học trên diện rộng. Owen và cộng sự (1992) đã nghiên cứu khả năng trợ giúp của computer để so sánh kết quả thu được khi nghiên cứu các chủng Helicobacter pylori. Tương tự như vậy Bialkowska-Hobranska và cộng sự (1990) dùng thiết bị laze đo mật độ các mảnh ADN lai cùng với sự trợ giúp của computer để phân tích các kết quả thu được từ các chủng cầu khuẩn nha bào gram âm. Phương pháp này có thể đưa ra số liệu chung làm cơ sở cho xác định sự giống nhau giữa các loài khác nhau. Các phân tích thu được từ các số liệu của các các thể khác nhau có thể chỉ ra được các cá thể mới làm cơ sở cho định danh cũng như theo dõi. Mọi nghiên cứu cho thấy một điều quan trọng và có ý nghĩa nhất là cách chọn enzym cắt hạn chế và loại mẫu dò dùng cho phép lai (Saunder và cộng sự 1991). Tóm tắt:Nhìn chung phương pháp lai ADN chứng tỏ ưu thế của nó như là một kỹ thuật quan trọng cho định typ và nghiên cứu dịch tễ học nhiều loại vi sinh vật khác nhau. Về nguyên tắc phương pháp này cũng có ưu điểm và nhược điểm của nó. Ưu điểm:- Sử dụng cho nhiều đối tượng vi sinh vật. - Có các mẫu dò vạn năng được bán rộng rãi trên thị trường. Kết quả lai có độ lặp lại cao và khá đơn giản cho việc phân tích kết quả. - Có thể sử dụng sự trợ giúp của computer để lưu giữ và phân tích kết quả thí nghiệm. Hạn chế: - Phương pháp sử dụng khá phức tạp và tốn thời gian. - Thông tin kết quả chỉ phản ánh cho đoạn gene lai với mẫu dò sử dụng. Hiện nay, có thể sử dụng các đoạn ADN tổng hợp làm mẫu dò và điều này thực tế đã làm cải thiện nhiều mặt của kỹ thuật lai như: khi có ADN tổng hợp thì không phải thực hiện các kỹ thuật tách và tinh sạch các mảnh ADN tách dòng có thể lẫn ADN plasmid. Mặt khác khi lai với mẫu dò tổng hợp thì phản ứng tiến hành nhanh với độ đặc hiệu cao hơn. Cho dù kỹ thuật này được sử dụng tốt cho nhiều đối tượng vi sinh vật khác nhau nhưng năm 1992 Heimberger và cộng sự đã tiến hành phân tích ADN được xử lý với enzym cắt hạn chế trong trường xung điện (PFGE), kết quả này rất có ý nghĩa cho phép phân tích sâu hơn và phân biệt các chủng vi sinh vật liên quan đến sự bùng phát dịch đối với các chủng vi sinh vật khác. Ribotyping và PFGE có chung nguyên tắc dựa vào sự phân bố của các vị trí enzym cắt hạn chế trên ADN của ribosom. Tuy nhiên, sự khác biệt ở chỗ ribotyping phản ánh sự phân bố của vị trí các enzym cắt hạn chế nằm trong các gene mã hoá cho rRNA hay nằm trong vùng nhiễm sắc thể có độ bảo thủ cao, còn đối với PFGE phản ánh sự phân bố của các enzym cắt hạn chế phân bố trên toàn bộ gene nghiên cứu. Nghiên cứu của Prevost (1992) cho thấy là kỹ thuật PFGE có thể hiệu quả hơn kỹ thuật ribotyping đối với phép phân tích các chủng Staphylococus aureus kháng methicillin.
2.3. Nhân gene và kỹ thuật giải trình tự ADN.Kỹ thuật lai ADN đã được sử dụng rộng rãi cho các nghiên cứu xác định typ của nhiều đối tượng vi sinh vật. Nguồn ADN đích đôi khi chỉ có số lượng ít trừ khi các tiến hành nuôi cấy vi sinh vật. Trong một số trường hợp việc nuôi cấy không thể thực hiện được với nhiều loài vi sinh vật hay virút hoặc trong các trường hợp khác cần thời gian dài nuôi cấy vi sinh vật để có đủ ADN cho các kỹ thuật lai hay định typ. Khó khăn này được khắc phục bằng cách làm tăng nguồn ADN đích một cách nhanh chóng bằng kỹ thuật nhân gene PCR. a. Kỹ thuật nhân gene PCR (Polymerase Chain Reaction)Việc nhân ADN cho phép làm tăng số lượng gấp hàng triệu hoặc nhiều hơn nữa đối với đoạn ADN hay ARN nghiên cứu. Có nhiều phương pháp khác nhau để phân tích nguồn ADN khuyếch đại theo cách này. Phương pháp đơn giản nhất là sử dụng điện di để xác định kích thước sản phẩm của phản ứng PCR. Có thể xác định độ nhạy và tính đặc hiệu cao hơn nếu sản phẩm PCR được lai với mẫu dò đặc hiệu đã được đánh dấu. Phương pháp đưa lại thông tin nhiều nhất là xác định được trình tự ADN và ARN của sản phẩm PCR. Việc xác định được sai khác của chuỗi ADN sẽ cho phép xác định và định typ chính xác nhất các đối tượng vi sinh vật nghiên cứu. Kết quả xác định trình tự ADN còn cho phép xác định mối liên hệ tiến hoá và dịch tễ học của các chủng vi sinh vật. - Đôi nét về phản ứng chuỗi PCR:Phản ứng chuỗi PCR nhằm khuyếch đại ADN lần đầu tiên được Saki (1985) giới thiệu và đây được coi là một trong những tiến bộ khoa học quan trọng nhất về sinh học trong thập kỷ 80. Phản ứng PCR sử dụng enzym chịu nhiệt tạo ra nhiều phiên bản theo hàm số mũ. Có thể ví dụ, chỉ cần 1 sợi ADN sau vài giờ có thể nhân lên 1011 phiên bản ADN tương đương với 100 ng ADN. Sau khi thực hiện phản ứng PCR thì cần tiến hành một số kỹ thuật để xác định sản phẩm, đơn giản nhất là điện di và xác định kích thước của sản phẩm PCR. Trong các nghiên cứu chẩn đoán thì kết quả này rất có ý nghĩa, nó chứng tỏ sự có mặt của ADN đích trong mẫu nghiên cứu. Có thể nhận được tính đặc hiệu và độ nhạy cao hơn khi sử dụng kỹ thuật RFLP với sản phẩm PCR hoặc lai với mẫu dò đặc hiệu. Tuy nhiên, thông tin đầy đủ nhất vẫn là kết quả xác định trình tự ADN của sản phẩm PCR. Ngay từ khi được giới thiệu thì PCR đã được xem là phương pháp đưa lại kết quả khá nhạy trong việc xác định và phát hiện vi sinh vật. Tính ưu việt của nó thể hiện ở chỗ có thể chỉ cần một hạt virus hay một tế bào vi khuẩn nào đó cũng đủ cho phản ứng xảy ra và khuyếch đại tới mức có thể phát hiện được trong thời gian ngắn. Kỹ thuật PCR còn được dùng để nhân ADN từ khuôn RNA sau khi đã được chuyển thành cDNA sau phản ứng phiên mã ngược (RT). Phương pháp này nhanh chóng được chú ý và trở lên phổ biến cho các nghiên cứu phát hiện vi khuẩn hay virus ở nồng độ thấp trong các mẫu môi trường và bệnh phẩm. Ví dụ việc phát hiện mẫu máu nhiễm HIV có nghĩa là phát hiện phần nhỏ ADN của virus trong hệ gene người có kích thước gấp 100 000 lần. Mặt khác khi dùng kỹ thuật miễn dịch với kháng thể thì phải cần tới 8 ngày mới có đáp ứng miễn dịch trong máu, trong khi đó với kỹ thuật PCR thì toàn bộ quy trình phát hiện chỉ mất vài giờ (đối với nguồn ADN hay RNA). Lợi thế của kỹ thuật PCR được tận dụng cho việc chuẩn bị ADN làm mẫu dò với số lượng lớn cho các phép lai ADN đã trình bày ở trên. Các mẫu dò có thể được đánh dấu bằng phóng xạ hay phi phóng xạ khi tiến hành phản ứng khuyếch đại. Mẫu dò đánh dấu với kỹ thuật PCR có ưu thế hơn các phương pháp đánh dấu truyền thống bằng kỹ thuật tổng hợp các mạch đứt gãy (nick translation) hay đánh dấu với mồi ngẫu nhiên (rADN random primer labeling). Các ưu thế này có thể kể đến, cụ thể như là cần lượng nhỏ sợi ADN khuôn, có thể tiến hành đánh dấu với các mẫu dò có kích thước ngắn, chuẩn bị mẫu dò không cần tinh sạch ADN khuôn. Bảng 1.7. dưới đây liệt kê một số ví dụ ứng dụng kỹ thuật PCR cho phát hiện nhiều đối tượng vi sinh vật khác nhau. Rất nhiều các kết quả nghiên cứu như vậy được trình bày trong các tạp chí chuyên ngành như: ành như: ành như: ành như: Journal of Clinical Microbiology, Applied Environmental Microbiology. Bảng 1.7. Một số ví dụ về ứng dụng kỹ thuật PCR trong xác định vi sinh vât.
Sau đây là những nét chung nhất của kỹ thuật PCR:Nguyên tắc cơ bản của PCR:+ Khái quát chung:PCR là phương pháp dùng enzym khuyếch đại chọn lọc một đoạn ADN theo luật số mũ. Phản ứng nhân gene được thực hiện với enzym ADN chịu nhiệt, hai mồi tổng hợp và 4 loại deoxyribonucleic (dATP, dGTP, dCTP và dTTP). Mỗi một chu kỳ phản ứng nhân gene gồm 3 bước (xem hình vẽ minh hoạ): Bước 1 là biến tính ADN, có tác dụng tách hai sợi đơn từ sợi khuôn xoắn kép. Bước 2 là bắt cặp hai mồi vào hai sợi đơn của khuôn. Bước 3 là kéo dài chuỗi theo mồi, chiều 5’-3’ với quá trình trùng hợp gắn các dNTP dọc theo sợi khuôn để tạo thành phiên bản mới theo nguyên tắc bổ sung. Tính đặc hiệu của phản ứng được quy định bởi mồi và sự sao chép trực tiếp theo trình tự sợi khuôn giữa hai mồi. Như vậy, kết quả tạo ra hàng triệu phiên bản sợi khuôn trong vài giờ. Mồi cho phản ứng là các đoạn ADN có kích thước 20-30 nucleotid được thiết kế theo trình tự của đoạn gene cần khuyếch đại dựa theo dữ liệu có sẵn. Mỗi cặp mồi phải hoàn thành chức năng của mình trong toàn bộ phản ứng, tức là chúng phải bám đặc hiệu vào đúng vị trí riêng cho từng mồi. Sở dĩ như vậy là do nếu chúng không bám vào vị trí đặc hiệu thì không thể khuyếch đại được đoạn gene đích. Với các gene có độ bảo thủ cao như rADN thì khi cặp mồi được thiết kế thì chúng có thể nhân được gene từ nhiều đối tượng. Ngược lại, trong nhiều trường hợp dùng mồi không đặc hiệu thì có thể nhân được các sản phẩm PCR không đặc hiệu. Tuy nhiên, cả hai kết quả trên đều được dùng cho định typ vi sinh vật. Thí nghiệm phản ứng PCR lần đầu tiên dùng mảnh Klenow của enzym ADN polymerase I từ E.coli. Tuy nhiên, enzym này bị biến tính tại 94oC và như vậy cần phải bổ sung enzym mới sau mỗi chu kỳ phản ứng. Đến nay quá trình này đã được cải thiện do dùng enzym Taq ADN polymerase (Taq = Thermus aquaticus ) chịu nhiệt. Enzym này được tách ra từ vi khuẩn sống ở suối nước nóng. Tốc độ xúc tác cho phản ứng của enzym này rất nhanh, có thể đạt khoảng 8000 bp/phút tại 755oC. Hoạt tính enzym giảm một nửa khi xử lý tại 92.5oC trong 230 phút hoặc 40 phút tại 95oC. Như vậy khi dùng enzym này thì không cần bổ sung enzym mới sau mỗi chu kỳ phản ứng.
+ Các thông số kỹ thuật:
Bất kỳ một phản ứng PCR nào cũng bắt đầu bằng việc tách ADN làm sợi khuôn. Nói chung, theo lý thuyết chỉ cần một sợi khuôn cũng đủ cho phản ứng tạo ra sản phẩm nhưng trong thực tế cần khoảng 10-100 ng/phản ứng. Đôi khi chuẩn bị ADN theo phương pháp đơn giản không cần tinh sạch cũng phù hợp cho phản ứng PCR. Tuy nhiên, điều quan trọng là tránh sự có mặt của EDTA (acide éthylène-diamine-tétraacétique) hoặc đệm phosphat vì kết quả sẽ làm giảm Mg++ (do EDTA hoặc tạo thành muối Mg3(PO4)2 khi nhiệt độ cao). Phản ứng PCR có thể nhân được đoạn gene dài 10 Kb nhưng trong thực tế thường dùng để nhân các đoạn có kích thước ngắn hơn (<2 Kb). Có thể tham khảo các thông số thành phần phản ứng PCR trong bảng dưới đây.
Bảng 1.8. Thành phần phản ứng PCR.
Số chu kỳ cho phản ứng tạo sản phẩm đặc hiệu từ 20-30 chu kỳ. Điều chú ý là các mồi cho phản ứng phải có nhiệt độ bắt cặp với ADN đích gần nhau, có trình tự không bổ sung nhau để tránh bắt cặp với nhau. Trình tự đầu 3’ của các mồi cũng phải khác với trình tự vùng trung tâm của sản phẩm để tránh tạo thành hiện tượng kẹp tóc. Nói chung, ngày nay người ta có thể tối ưu hoá việc chọn mồi nhờ sự trợ giúp của các chương trình máy tính. Đôi khi cũng xuất hiện hiện tượng nhân sản phẩm PCR không đặc hiệu, điều này thường hay xuất hiện khi dùng các cặp mồi suy biến từ chuỗi acid amin. Tuy nhiên trong hầu hết các trường hợp này thì việc thay đổi điều kiện phản ứng có thể hạn chế được các sản phẩm không mong muốn. Các thành phần như nhiệt độ, nồng độ Mg++ và enzym có ảnh hưởng lớn đến tính đặc hiệu. Sự có mặt của nonidet-40 có tác dụng tăng hiệu suất của phản ứng. Việc ứng dụng kỹ thuật RFLP với sản phẩm PCR có thể là cách nhanh chóng để tìm sự khác biệt đối với các gene khác nhau. Với cách này yêu cầu sản phẩm PCR phải sạch và đồng nhất. Theo cách này trình tự có thể được thực hiện như sau: Sau khi điện di kiểm tra độ sạch của sản phẩm PCR, tiến hành kết tủa với ethanol. Kết tủa được hoà với đệm thích hợp và sau đó được xử lý với enzym cắt hạn chế thích hợp. Kiểm tra kết quả thông thường trên gel agarose, trong một số trường hợp có thể trên gel polyacylamid để thu được độ phân giải cao hơn. Chú ý rằng thông thường hoạt tính enzym giảm 5% sau mỗi chu kỳ nhiệt. Như vậy có thể ước lượng 50% hiệu quả khuyếch đại mất đi trong toàn bộ quá trình phản ứng. + Một số khó khăn với phản ứng PCR:
PCR là kỹ thuật có ý nghĩa lớn trong nghiên cứu sinh học phân tử nhưng cũng nảy sinh một số vấn đề cần chú ý đặc biệt khi phát hiện một số bệnh virus hay vi khuẩn. Vấn đề ở đây là acid nucleic của cả tế bào chết và tế bào sống đều cho kết quả dương tính trong phản ứng PCR. Phản ứng nhạy nên mọi sự nhiễm ADN đều có thể đưa đến sản phẩm dương tính giả trong kết quả. Để hạn chế thực tế này cần tiến hành với các mẫu đối chứng cần thiết để biết được sự nhiễm tạp từ các thành phần phản ứng hay từ dụng cụ thí nghiệm.
Một vấn đề nữa cũng cần đề cập là sai số trong phản ứng nhân gene với enzym taq ADN polymerase. Sai sót thêm nucleotid xảy ra với mỗi một đơn vị sao chép khoảng 9000 nucleotid và đột biến dịch khung xảy ra với số lượng tương ứng khoảng 40 000 nucleotid. + Một số kỹ thuật PCR khác:Một số kỹ thuật phát triển trên cơ sở của kỹ thuật PCR có ý nghĩa quan trọng cho định danh và định typ vi sinh vật là nested PCR (PCR lồng), revert transtriptase PCR (sao chép ngược) và asymmetric PCR (PCR một chiều). PCR lồng là phản ứng PCR bao bồm hai phản ứng đồng thời, phản ứng đầu ở vòng ngoài gene và sản phẩm của nó lại làm khuôn cho phản ứng sau cho đoạn nằm trong gene. Như vậy phản ứng sau cần 1 hay 2 mồi để nhân phần gene nằm trong sản phẩm PCR từ hai mồi phía ngoài tạo ra. Vai trò kỹ thuật này làm tăng độ nhạy và đặc hiệu của phản ứng PCR. Phản ứng PCR phiên mã ngược được dùng để nhân ADN từ ARN của virus. Việc kết hợp phản ứng này với PCR lồng có tác dụng phát hiện ARN tại thời điểm nhất định của mẫu. Kỹ thuật PCR một chiều được ứng dụng trong phương pháp giải trình tự ADN trực tiếp từ sản phẩm PCR.phẩm PCR. + Sử dụng kỹ thuật PCR cho xác định typ vi sinh vật:Như đã trình bày ở trên, dùng cặp mồi đặc hiệu có thể xác định được gene đặc trưng cho loài hay thậm chí một chủng vi sinh vật nào đó. Các chiến lược ứng dụng lợi thế của kỹ thuật PCR dùng trong xác định typ vi sinh vật trong nghiên cứu dịch tễ học được chia làm 4 nhóm sau: - Phân tích RFLPs với sản phẩm PCR khi dùng cặp mồi đặc hiệu.Trong trường hợp này dùng kết hợp kỹ thuật PCR và xử lý sản phẩm PCR với enzym cắt hạn chế và quan sát kết quả sau khi điện di sản phẩm trên gel agarose hoặc polyacrylamid Một dẫn chứng cho kỹ thuật này là kết quả nhân gene synthase citrat trong Ricketsiae sau đó sản phẩm PCR được xử lý với enzym cắt hạn chế để phân biệt giữa các chủng với nhau (Regnery và Ctv, 1991). Các ví dụ sau (bảng 1.8) đã cho thấy hiệu quả của phương pháp này. Bảng 1.9. Một số ví dụ cho kỹ thuật RFLPs (Restriction Fragment Length Polymorphism) phân tích các gene đặc hiệu được nhân lên bằng kỹ thuật PCR.
+ Dùng kỹ thuật PCR cho ribotyping:Kỹ thuật PCR ribotyping là ứng dụng các cặp mồi đặc hiệu để nhân các vùng gene của ARN (thuộc ribosom hay ARN vận chuyển). Tuy nhiên, trong thực tế kỹ thuật này yêu cầu kinh nghiệm, kỹ thuật và tốn thời gian để thực hiện phép lai. Để khắc phục hạn chế này một cách tiếp cận được trình bày ở đây là phân tích đa hình (dấu vân tay) của vùng gene này hay vùng gene xen kẽ của rARN hoặc tARN. Hầu hết các chủng vi khuẩn đều chứa 2-22 phiên bản rARN trong mỗi tế bào (Gottlie và Rundner, 1985) trong khi đó vùng xen kẽ giữa 16S và 23S chứa một số các đoạn ADN lặp lại (Bacot và Reeves, 1991). Trong khi đoạn gene mã hoá cho tARN khá bảo thủ thì vùng xen kẽ của tARN lại thay đổi từ 2-35bp đối với Bacillus subtilis (Vod, 1985) và từ 2-208bp trên đối tượng E.coli (Jinks-Roberson và Nomura, 1987). Như vậy khi sử dụng cặp mồi nhân toàn bộ vùng gene tARN có thể tạo ra được sự khác biệt giữa các chủng vi sinh vật. Kết quả ứng dụng kỹ thuật này trên một số đối tượng được trình bày trong bảng 1.9.
Bảng 1.10.. Một số ví dụ sử dụng kỹ thuật PCR cho ribotyping.
Khi số phiên bản rARN tăng có nghĩa là làm tăng độ nhạy của phương pháp ứng dụng. Engstrand và cộng tác viên (1920) đã công bố có thể sử dụng mẫu dò là đoạn nucleotid bổ sung với đoạn 16S của rARN như là một trong số mồi cho phép nhân phiên mã ngược ARN và phép phân tích được thực hiện nhờ kỹ thuật PCR nhân sản phẩm cDNA được dùng cho phát hiện Helicobacter spp. Độ nhạy của phương pháp phát hiện này tăng 50 lần so với phương pháp truyền thống. + Kỹ thuật rep-PCR:Một phương pháp trực tiếp cho kết quả finger printing không cần sử dụng enzym cắt hạn chế là rep-PCR dựa trên nguyên tắc nhân các đoạn gene lặp lại. Thuật ngữ rep-PCR là chỉ phương pháp chung sử dụng cặp mồi đặc hiệu để nhân các chuỗi lặp lại phổ biến trong các vi sinh vật thuộc cơ thể nhân sơ. Các chuỗi lặp lại có độ bảo thủ cao trong các đối tượng thuộc Enterobacteriaceae và một số đối tượng khác (Versalovic và Lupsky, 1991). Dùng các đoạn mẫu dò bảo thủ có thể lai với cả hai đoạn ADN lặp lại: đoạn có kích thước 126bp (Enterobacterial repetitive intergeneic concensus-ERIC) và đoạn 38bp (repetitive extrageneic palindromic-REP) từ bộ gene của Enterobacteriaceae vi khuẩn gram âm và một số chủng có quan hệ xa khác. Có thể trực tiếp thiết kế cặp mồi cho các đoạn gene bảo thủ để thu được kết quả finger printing sau khi nhân gene dùng bộ gene của các vi sinh vật có mang các đoạn gene bảo thủ này. Sau khi điện di có thể phát hiện được trên agarose các đoạn gene có kích thước khác nhau từ các nguồn vi sinh vật khác nhau. Kỹ thuật này được ứng dụng đã đem lại kết quả tốt khi nghiên cứu mối quan hệ trên các đối tượng Bacillus subtilis (Versalovic và cộng sự, 1992), Citrobacter diversus (Woods và cộng sự, 1991), Rhizobium meliloti (Brujin, 1992). Thực tế phương pháp tạo ra kết quả finger printing đa dạng từ hầu hết các đại diện của Enterobacteriaceae (Versalovic và cộng sự, 1991) và được ứng dụng trên tất các vi khuẩn có mang các đoạn gene bảo thủ lặp lại trên. Phương pháp tương tự cũng cho phép phát hiện sự khác nhau từ các nguồn ADN trên các đối tượng nhân thực thuộc chi Naegleria như mô tả của Belkim và Quint (1992). Phương pháp này dùng cho phát hiện đa hình các đoạn gene chung cho cả vi sinh vật nhân sơ và nhân chuẩn. + Kỹ thuật RAPD (Random amplified of polymorphic DNA):Hạn chế của các phương pháp nghiên cứu trước đây là phải dùng cặp mồi đặc hiệu. Theo cách này phải có các thông tin liên quan đến đoạn gene nhất định và đối tượng nghiên cứu. Khó khăn này được loại bỏ trong trường hợp dùng các mồi ngẫu nhiên hay tuỳ tiện, như vậy phương pháp phân tích không cần đến thông tin về geneome của đối tượng nghiên cứu…Cơ sở của phương pháp này đã được Welsh và McClell ADN (1990) mô tả khi dùng mồi ngẫu nhiên nhân gene và tạo ra phổ finger printing đặc trưng cho từng hệ gene (geneome). Mồi ngẫu nhiên có thể chỉ gồm 5 bp để nhân gene có thể tạo ra kết quả finger printing khá đa dạng. Có thể xem kết quả minh hoạ trong bảng và hình sau. Bảng 1.11. Ví dụ minh hoạ cho kỹ thuật RAPD.
Hình 1.9. Minh hoạ kết quả phân tích sử dụng kỹ thuật RADP. b. Giải trình tự ADNPhương pháp chính xác và đưa lại nhiều thông tin nhất cho định danh và định typ vi sinh vật là xác định chính xác trình tự chuỗi ADN của một vùng quy định trên nhiễm sắc thể. Phương pháp giải trình tự ADN phát triển nhanh chóng, kết quả so sánh sự tương đồng của trình tự gene nghiên cứu trở thành phương pháp phân loại chuẩn theo sinh học phân tử trong nghiên cứu hệ thống học và cây phả hệ di truyền giữa các đối tượng nghiên cứu. Các gene bảo thủ được giải trình tự làm cơ sở để xác định vị trí của sinh vật nghiên cứu, trong khi đó các trình tự không tương đồng (khác biệt) lại làm cơ sở cho sự biệt hóa cũng như xác định mối quan hệ giữa các cá thể gần với nhau. + Nguyên tắc:Phương pháp giải trình tự ADN theo nguyên tắc tạo ra các mảnh ADN được đánh dấu tại đầu 5’ hoặc 3’. Các mảnh có các base đánh đấu được tách ra trên gel polyacrylamid Các mảnh được phân biệt về vị trí trên gel với sự sai khác chỉ với một nucleotid. Việc đánh dấu và đọc kết quả theo các kỹ thuật và phương pháp khác nhau. Việc tạo ra các mảnh ADN sai khác nhau về 1 nucleotid được trình bày theo 2 phương pháp: phương pháp hóa học (Maxam & Gilbert, 1997) và phương pháp enzym kết thúc phản ứng chuỗi (Sanger, 1997). Ngày nay phương pháp enzym được dùng phổ biến hơn cả. + Phương pháp enzym kết thúc phản ứng chuỗi của Sanger:Phương pháp kết thúc phản ứng chuỗi được thực hiện dựa trên việc nhân ADN từ sợi khuôn khi có đoạn ADN mồi với xúc tác của Klenow hay T7-ADN polymerase và 4 loại deoxyribonucleotid (dNTPs). Như vậy, có 4 phản ứng được tiến hành đồng thời và trong mỗi phản ứng thì có một loại dNTP bị thay đổi bởi một dẫn xuất để làm dừng phản ứng (ddNTP). Kết quả là phản ứng chuỗi bị dừng đặc hiệu cho mỗi một base xác định xảy ra một cách ngẫu nhiên trong mỗi phản ứng. Trình tự của ADN được xác định trên gel. Đầu tiên phương pháp này được xây dựng cho xác định trình tự sợi ADN đơn tạo ra bằng cách tách dòng ADN vào vectơ thực khuẩn thể M13 (Sanger và cộng sự, 1978). Sau đó phương pháp xác định trình tự sợi đôi ADN đã được Chen, William (1986) mô tả khi tách dòng ADN vào plasmid. Ngày nay người ta lựa chọn phương pháp Sanger để giải trình tự ADN cho nghiên cứu phân loại và xác định typ vi sinh vật thay cho phương pháp hóa học. Một số lợi thế của phương pháp này ở chỗ: Phương pháp này đơn giản và không tốn nhiều thời gian như phương pháp hóa học. Khi nghiên cứu phân loại và xác định typ thì các gene nghiên cứu có độ bảo thủ nhất định vì vậy ta có thể dùng chung mồi cho nhiều đối tượng khác nhau. Một trong hạn chế của phương pháp này ở chỗ các đoạn ADN cần được tách dòng trước khi tiến hành giải trình tự ADN. Tuy nhiên cho đến nay người ta đã cải tiến phương pháp này để có thể giải trình tự trực tiếp sản phẩm PCR. + Giới thiệu kỹ thuật giải trình tự ADN theo Sanger:Có hai phương pháp: Phương pháp giải trình tự truyền thống đọc kết quả trên bản gel (đánh dấu bằng phóng xạ là chủ yếu) và phương pháp giải trình tự và đọc kết quả tự động (đánh dấu bằng hóa chất huỳnh quang). - Phương pháp truyền thống: Các bước chính cho giải trình tự ADN như sau: Chuẩn bị ADN khuôn Bắt cặp ADN khuôn và mồi Tiến hành phản ứng giải trình tự ADN Tách sản phẩm trên gel polyacrylamid biến tính với urea. Đọc kết quả. Tuy nhiên cần chú ý một số điểm sau: 1. Chuẩn bị ADN khuôn. 2. Điều kiện bắt cặp mồi. 3. Thực hiện phản ứng giải trình tự ADN. 4. Tách sản phẩm trên acrylamid gel. 5. Đọc kết quả. Một số điểm cần được chi tiết thêm cho mỗi bước như sau:1, Chuẩn bị ADN khuôn:Sợi khuôn cần được hiểu là việc giải trình tự được thực hiện với sợi đơn (vectơ M13) hay sợi đôi ADN. Việc đọc trình tự theo sợi đơn thường cho kết quả là kích thước đoạn gene đọc dài và độ chính xác cao. Ngày nay người ta thường đọc sợi đôi vì việc chuẩn bị mẫu dễ dàng hơn, ngoài ra khi sử dụng hai phản ứng với hai mồi tương thích thì quá trình đọc lại chính xác hơn. Đó là ưu thế của cách đọc sợi đôi ADN. Do vậy, rất cần đọc kết quả từ cả hai sợi ADN. Hiện nay phương pháp đọc trình tự ADN trực tiếp từ sản phẩm PCR đang ngày càng trở lên phổ biến. 2, Chuẩn bị sợi ADN mồi cho bước bắt cặp vào sợi khuôn:Người ta có thể dùng các đoạn ADN mồi tiêu chuẩn đặc hiệu cho trình tự ADN của vectơ gần sát với sợi khuôn hay primer đặc hiệu của sợi khuôn mà nó sẽ bắt cặp vào. Tiện ích của việc dùng mồi chuẩn ở chỗ mọi điều kiện đã được lựa chọn cho sợi khuôn và mồi được dùng lặp đi lặp lại nhiều lần cho các sợi khuôn khác nhau. Hạn chế của loại mồi này ở chỗ chỉ đọc được một đoạn ngắn của gene (300 – 500 bps) trong trường hợp phải đọc các gene có kích thước lớn cần phải tiến hành subclone để đọc tiếp. Điều này có thể thực hiện bằng cách tạo ra các đoạn ADN ngẫu nhiên từ toàn bộ gene sau đó lại gắn vào vectơ để đọc cho hết trình tự của gene. Phương pháp tiếp theo là sử dụng mồi được thiết kế theo nguyên tắc bổ sung với trình tự của gene. Theo cách này, lần lượt các đoạn được đọc mà không cần thao tác subclone. Tuy nhiên theo cách đoạn mồi được thiết kế để đọc gene như vậy thì điều kiện cho từng phản ứng giải trình tự ADN với các mồi khác nhau sẽ không được tối ưu. Tuy vậy, hiện nay việc tổng hợp mồi đã trở nên dễ dàng và phổ biến với giá thành thấp, hơn thế nữa với sự trợ giúp của tin học thì bước thiết kế mồi đã được tối ưu hoá. Vì vậy phương pháp này ngày càng trở lên thông dụng hơn. Sau khi mồi đã được thiết kế thì việc tiếp theo là chọn cách đánh dấu vào mồi hay vào đoạn ADN sẽ được tổng hợp. Ưu thế của việc đánh dấu ngay tại đầu 5’ của mồi ở chỗ các băng ADN thường có cùng một cường độ tín hiệu. Thông thường với phương pháp đọc trình tự thủ công thì P32 thường được dùng. Ưu thế của nó là mang gốc photphat cao năng bêta mạnh, do đó chỉ cần trong thời gian ngắn là thu được tín hiệu. Hạn chế của phương pháp này là do mang năng lượng cao nên khó phân biệt giữa các băng khác nhau so với dùng S35. Hiện nay để giải quyết vấn đề này người ta dùng P33 tuy giá thành cao nhưng cho kết quả tốt hơn. 3, Thực hiện phản ứng giải trình tự ADN:Enzym thông dụng thường dùng là sequenase (là một dạng của T7 DNA polymerase), enzym này có ưu thế hơn hẳn so với Klenow được dùng trước đây. Phản ứng được thực hiện nhanh hơn và thu được kết quả tối ưu nếu có mặt ion Mn++, ion này giúp cho quá trình sao chép có độ chính xác cao. Trước đây dNTPs đánh dấu với P32 được dùng để đánh dấu sản phẩm. Tuy nhiên ngày nay nhiều người đã chuyển sang dùng S35 thay thế cho P32, kết quả là cho độ phân giải cao hơn. Mới đây phương pháp giải trình tự dựa trên kỹ thuật PCR được gọi là chu kỳ giải trình tự trực tiếp ADN đang thay thế phương pháp cũ dùng enzym sequenase. Về nguyên tắc thì các phương pháp này là giống nhau. Ưu thế của phương pháp này là: lượng ADN đòi hỏi ít và không yêu cầu phải có mức độ tinh sạch cao. Ngoài ra, có một ưu điểm nữa là phản ứng xảy ra ở nhiệt độ cao, do đó đã khắc phục được những hạn chế của các phương pháp khác khi giải trình tự đối với các sợi khuôn có cấu trúc bậc 2 (Fan & cộng sự, tạp chí Biotechniques 21: 1132-1137). 4, Tách sản phẩm trên gel urea-acrylamid:Cho đến nay đã có nhiều tiến bộ trong kỹ thuật điện di gel kể từ năm 1970 khi mà người ta phát hiện ra phương pháp này. Tiến bộ quan trọng nhất là gel gradient tạo ra độ dãn cách thích hợp để đọc được thêm từ 50-100 base. Đầu tiên, gradient được tạo ra giữa acrylamid và gradient muối khi đổ gel. Có thể có cách khác nữa là người ta thay đổi độ dày của gel bằng spacer. Phương pháp dễ nhất là bổ sung nồng độ muối NaOAc vào bể đệm dưới khi chạy gel, kết quả sẽ tạo ra một gradient cần thiết mà không mấy khó khăn. Có nhiều cách cải tiến khác nhau để tạo ra gel có độ phân giải cao, ví dụ tạo ra gradient đối với acrylamid mạch thẳng hay bổ sung một số chất tạo ra gradient (Long Ranger). Mặt khác việc đổ gel cũng được đơn giản hóa bước sử dụng băng dán. Một số người dùng các gel siêu mỏng để đạt độ phân giải cao. Nhiều thiết bị được phát minh nhằm thu nhận các băng đã được tách riêng khi chúng ra khỏi gel, và gần đây những thiết bị này đã được thương mại hóa. Nhuộm bạc là phương pháp tương đối hiệu quả để xác định các trình tự ADN, cách này hạn chế được nhiều bước dùng trong các kỹ thuật dựa vào ảnh bức xạ mà không cần sử dụng chất phóng xạ. 5, Đọc trình tự gene:Hiện nay kỹ thuật đọc phim tự động đã thay thế công việc đọc trình tự ADN buồn tẻ trước đây. Có hàng loạt các thiết bị từ loại có thể ghi lại trình tự dựa vào việc chọn từng loại base đến những thiết bị có thể thực hiện được toàn bộ quá trình này một cách tự động. Hy vọng rằng với những bàn luận ngắn gọn này sẽ giúp bạn làm quen dần với nhiều loại kỹ thuật khác nhau. Ngày nay các thiết bị giải trình tự tự động càng trở lên thông dụng và việc sử dụng loại thiết bị và hóa chất khác nhau rất đa dạng. Việc chọn lựa thiết bị, hóa chất và phương pháp thích hợp để giải trình tự ADN nhanh, chính xác và kinh tế là điều cần được quan tâm đến.
* Một số phương pháp cụ thể thường được dùng cho kỹ thuật giải trình tự ADN.Phương pháp 1: Tinh sạch Plasmid cho giải trình tự ADN.Hiện nay có nhiều kỹ thuật tinh sạch plasmid khác nhau, phương pháp được trình bày dưới đây là phương pháp thu được plasmid có độ tinh sạch cao, có thể dùng cho cả giải trình tự ADN thủ công và tự động hóa. Quá trình này là cải tiến phương pháp sử dụng dịch kiềm làm tan tế bào, sau đó sử dụng sắc ký qua cột trao đổi ion. Cũng như các sản phẩm được lưu hành trên thị trường của các công ty, hạn chế lớn nhất của phương pháp này là giá thành của nhựa trao đổi ion tương đối đắt. Thực tế có rất nhiều công việc phải giải trình tự một cách thủ công và các cán bộ nghiên cứu đã tìm ra nhiều phương pháp tinh sạch khác nhau để thu nhận ADN đạt độ sạch cần thiết, thậm chí có thể dùng cho giải trình tự ADN tự động. Tuy vậy, nhiều người vẫn thích sử dụng cột trao đổi ion, kết quả có thể đọc được tới 600 bps hoặc nhiều hơn nữa. Quá trình tinh sạch như sau: + Nuôi cấy vi khuẩn mang plasmid trên môi trường chọn lọc với kháng sinh tương ứng (khoảng 3 ml).1, Phân 3 ml môi trường LB chứa 100 mg/ml apicilin trong ống nghiệm vô trùng (12X100 nm có nút). 2, Vô trùng đầu que cấy trên lửa và làm nguội trên mặt đĩa thạch. 3, Lấy sinh khối từ một khuẩn lạc và đưa vào ống nghiệm chứa môi trường nuôi cấy trên. 4, Nuôi cấy vi khuẩn 37oC qua đêm có lắc vừa phải. + Chuẩn bị ADN dùng cột Qiagene:Các cột Qiagene được chuẩn bị dễ dàng và nhanh để tinh sạch ADN không cần dùng gradient CsCl. ADN được chuẩn bị theo phương pháp này đạt kết quả rất tốt cho việc biến nạp và giải trình tự gene. Có hai chú ý quan trọng khi dùng cột Quiagene là: (1) Không sử dụng quá nhiều mẫu lên cột, điều này rất quan trọng vì nếu dùng thể tích quá nhiều kết quả là sẽ có nhiều protein ảnh hưởng đến khả năng bám cột của ADN. Với cách này có thể nhận được lượng ADN có chất lượng cao hơn từ thể tích ít hơn. Nếu như lượng ADN thu được ít từ lần thử đầu tiên thì cần thực hiện đúng theo chỉ dẫn để lần thử thứ 2 với thể tích lớn hơn. Khi có một loạt thể tích mẫu được Quiagene khuyến cáo lên cột để đạt được hiệu suất tinh sạch cao nhất thì nên dùng lượng dịch lên cột có thể tích nhỏ nhất. (2) Đảm bảo cột phải được rửa hai lần trước khi giải phóng ADN ra khỏi cột, điều này cũng rất có ý nghĩa vì việc rửa như vậy sẽ loại trừ được protein và các thành phần tạp nhiễm khác. + Các bước dùng cột trao đổi Qiagene (tài liệu hướng dẫn sử dụng Quiagene):1. Ly tâm khoảng 1,3 ml dịch vi khuẩn mang plasmid. Làm tan với 0.3 ml đệm P1 có RNase, chuyển sang tube mới. 2. Thêm vào 0.3 ml đệm P2 và trộn đều, giữ ở nhiệt độ phòng 5 phút (không nên để lâu hơn). 3. Thêm vào 0.3 ml đệm P3 trộn đều ngay nhưng phải nhẹ nhàng, ly tâm ở 4oC trong 30 phút với tốc độ 15000 v/phút. Thu lấy phần trên tủa. 4. Ly tâm lại như bước 3 một lần nữa để loại các phần cặn nếu có. 5. Cân bằng đệm cho cột Qiagene-tip 20 với 1ml QBT và để cho đệm chảy hết tự do. 6. Đưa phần dịch thu được ở bước 4 lên cột và để tự chảy qua cột. 7. Rửa cột 2 lần, mỗi lần 1 ml đệm QC. 8. Thu ADN với 0.8 ml đệm QF. 9. Kết tủa ADN với 0.7 thể tích isopropanol (trước đó đưa về nhiệt độ phòng). Ly tâm 15000 v/phút tại 4oC. 10. Rửa ADN thu được với ethanol 70%, để khô 5 phút và hoà tan lại trong thể tích đệm thích hợp (25ml). Chú ý trong trường hợp mẫu dùng cho giải trình tự ADN tự động huỳnh quang thì hoà tan mẫu trong nước. Nói chung sử dụng cột Qiagene thu được trên 90% ADN và thường được dùng cho tinh sạch Plasmid và cosmit (kích thước dưới 2 Kbs đến lớn hơn 50 Kbs). Số lượng plasmid thu nhận được từ tế bào E.coli phụ thuộc vào hệ tế bào chủ, loại plasmid từ thấp trung bình đến số lượng plamid cao và điều kiện nuôi cấy (có thể tham khảo tài liệu liên quan từ các chỉ dẫn của Qiagene để quyết định số lượng tế bào dùng để xử lý cho mỗi loại kích thước cột). Chú ý cách làm sạch và tái sử dụng cột: 1, Thêm ethanol đầy vào cột, ly tâm. 2, Bỏ ethanol và ly tâm lại một lần nữa. 3, Lặp lại bước 1 và 2 với nước cât. 4, Giữ cột ở trạng thái khô cho đến lần sử dụng về sau. (Cột có thể được dùng cho nhiều lần tuy nhiên khi muốn tinh sạch ADN dùng cho tách dòng thì nên dùng cột mới).
Các loại dịch đệm dùng cho cột Qiagene:
Phương pháp 2. Chuẩn bị gel cho giải trình tự ADNGiới thiệu chung: Phương pháp này giới thiệu cách đổ gel, lên mẫu và chạy gel giải trình tự ADN. Có nhiều cách đổ gel, có thể tham khảo các cách khác nhau để chọn cách tiện ích nhất. Điều quan trọng nhất là các bản kính phải được rửa sạch tuyệt đối. 1, Mang găng tay và loại bỏ hết bột găng tay đi. Rửa bản kính một lần với nước nóng. Sau đó dùng bàn chải rửa với 0.5N NaOH. Lau bản kính với dung dịch Alconox hay chất tẩy rửa có chức năng tương đương. Sau đó rửa sạch lại bằng nước. 2, Rửa các bản kính với nước trao đổi ion sau đó dựng lên giá cho khô, tránh bụi bám vào kính. Lau với ethanol và dùng giấy Kimwipes hay Scott Utinity để lau cho sạch không tạo vết. Kiểm tra lại bụi bám vào kính để lau cho sạch, sau đó kính được lau bằng lớp dịch Rain-X hay Sigmacott, Gel Slick của hãng AT Biochem. Mục đích dùng chất này là giúp cho việc đổ gel dễ dàng hơn và gel không bị dính vào các bản kính. 3, Rửa lại bản đệm cách gel (spacer) với nước đặt vào dọc theo hai cạnh đối xứng của bản kính. 4, Ghép hai bản kính với nhau. Đặt cẩn thận hai bản kính bằng nhau sao cho hai bản spacer sát tận cuối bản gene (tạo mặt phẳng cùng với hai bản kính). Nếu không đảm bảo mặt phẳng thì dễ bị chảy gel trong quá trình đổ gel. Kẹp 2 cạnh của gel tại 3 vị trí. 5, Chuẩn bị gel acrylamid 8% Long Ranger (Hoefer Rig). Trộn đều hỗn dịch: 16 ml Acrylamid Long Ranger (LR) 50%. 5 ml 10X TBE 70 ml 10M Urea (46 gam) Đưa đến thể tích 100 ml với nước Cho vào 800 μl 10% Ammonium persulphat. 50 μl TEMED. Quá trình polyme hóa được thực hiện trong 15 -20 phút. Chạy gel LR trong dải nhiệt độ 50-55oC, nhiệt độ cao hơn 55oC sẽ phá các liên kết acrylamid LR gel là dạng cải biến cho gel acrylamid có một số ưu điểm sau: Thứ nhất là LR gel với nồng độ cao hơn nhưng việc chạy gel thì cũng như gel có nồng độ thấp thông thường, ví dụ LR gel 5% tương đương 4% gel acrylamid với bisacrylamid Thứ 2 là LR gel có khả năng phân giải cao hơn và chạy với tốc độ nhanh hơn 30-40% gel thông thường, gel LR khô nhanh hơn và không bị dính. Một số chú ý khi thao tác::a. Trộn gel nhẹ nhàng (tránh trộn mạnh). b. Hút dịch acrylamid bằng pitpet hay có thể dùng cốc đong. c. Đặt bản kính nghiêng 15o và đổ gel bằng cốc đong hay pipet. Kiểm soát tốc độ đổ gel bằng góc nghiêng. Tránh để dòng gel khi đổ ngắt quãng vì điều này sẽ dẫn đến tạo bọt. Nếu phải dừng lại để hút dịch mới vào syringer thì lúc đó phải hạ dần bản kính xuống vị trí nằm ngang trước khi dừng đổ gel. Sau đó lấy dịch gel mới vào syringer và tiếp tục đổ gel trước khi gel lên khỏi vị trí nằm ngang. Cứ thao tác như vậy đến khi đổ đầy bản gel. Nếu xuất hiện bọt khi đổ gel thì phải dừng lại ngay và nghiêng tấm kính để cho bọt khí đi lên, có thể gõ nhẹ tay vào bản kính để cho bọt khí đi lên hết. Phải loại hết bọt khí ra khỏi gel, sau đó mới tiếp tục đổ gel. 6, Đặt gel nằm ngang và đưa cạnh bằng của lược răng cá mập vào gel, tạo một góc hơi nghiêng nhằm tránh tạo bọt khí. Chắc chắn rằng đã đặt cạnh bằng của lược vào phía trên của gel và phần răng cá mập sẽ nằm dọc theo đỉnh của bản kính dài (lược sẽ nằm ngược với vị trí trong hình 1.10). Bổ sung thêm gel và đẩy thêm lược vào đúng vị trí và cố định lược lại bằng kẹp để tránh xê dịch. 7, Bao bọc gel với giấy nilon SaranWrap để tránh gel tiếp xúc với không khí. Ngoài ra có thể thấm giấy lọc với đệm TBE để đậy lên. Điều này giữ cho gel luôn ẩm và tránh gel tiếp xúc với oxy (oxy làm cản trở quá trình polyme hóa). 8, Quá trình polyme xảy ra từ 30 phút đến 1 giờ. Để xác định polyme hóa có thể hút một ít gel vào pipét pastuer và để cho đến khi quá trình polyme hóa xảy ra. Hoặc có thể lấy gel cho vào ống falcon và đậy chặt nắp lại, trong nhiều trường hợp thì gel chỉ nên polyme hóa trong 15-20 phút hoặc nhanh hơn. Nếu quá trình polyme hóa không xảy ra thì lại phải tiến hành rửa kính và đổ lại gel. Chú ý nhiều khả năng quá trình polyme hóa không xảy ra do Ammonium persulphat không còn tốt khi dùng lâu, nên chuẩn bị dịch này để dùng trong tuần. Điều cần chú ý nữa là gel phải được bọc kỹ với SaranWrap để qua đêm. 9, Chuẩn bị gel trước khi chạy: a. Lấy SaranWrap ra khỏi gel. b. Đặt gel vào thiết bị điện di (bản kính dài đặt áp vào thiết bị), chắc chắn là đã cắt bỏ lớp băng dính ở đáy gel để việc điện di xảy ra. c. Đổ 0,5X TBE vào bể đệm dưới của thiết bị, lượng đệm cần phải đủ để cho bản gel nằm trong đệm. d. Đổ đệm vào bể trên của thiết bị, đảm bảo gel nằm dưới lớp đệm. Kiểm tra xem đệm có bị chảy ra hay không. e. Chạy thử 20-30 phút tại hiệu điện thế 2000V (phải cẩn thận vì rất nguy hiểm), sau đó nhiệt độ của bản gel nên đạt tới 50oC. 10, Xử lý nhiệt với các mẫu ADN điện di tại 85oC trong 5 hút và trộn đều, để trên đá cho đến khi lên mẫu. 11, Tắt nguồn điện vào thiết bị điện di (nhớ ngắt nguồn điện vào thiết bị trong bất kỳ trường hợp nào khi thao tác với thiết bị). 12, Trong khi xử lý nhiệt với các mẫu thì rửa bề mặt với đệm trong bể điện di, dùng pipet hay syringer 60 ml. Mục đích của thao tác này là loại lớp urea trên bề mặt gel. 13, Đánh dấu vị trí đỉnh của mặt gel trên bản kính trước của gel. 14, Đặt lược răng cá mập lên mặt gel, các răng được đặt trên mặt gel và ngập sâu vào mặt gel khoảng 1 mm. Chắc chắn là vùng không gian giữa các răng được giới hạn giữa các răng và gel. Như vậy phần lược răng cá mập trở thành các cạnh bên của giếng, bề mặt gel là đáy của giếng tra mẫu (hình 1.10).
Hình 1.10. Vị trí đặt lược sau khi đổ gel.
Phải đảm bảo chắc chắn đã làm sạch bề mặt gel trước khi đặt lược răng cá mập lên trên gel. Nếu không thực hiện việc này sẽ không đọc được phần kết quả phía trên của gel. Nếu quyên thực hiện bước này có thể rửa sạch từng giếng tra mẫu nhưng việc này sẽ khó khăn hơn. Nếu lên nhiều mẫu thì trước đó cần phải phải làm sạch giếng vì urea sẽ khuyếch tán và tích tụ tại bề mặt gel tại thời điểm lên mẫu. 15, Việc xử lý nhiệt ở 85oC đối với các mẫu điện di nhằm làm biến tính ADN, chỉ lên khoảng 2-4 ml mẫu cho mỗi giếng (lên mẫu theo thứ tự ACGT), có thể dùng bút để viết lên kính nhằm tránh nhầm lẫn. 16, Quá trình điện di với hiệu điện thế 2000V cho đến khi màu BPB (Bromophenolblue) di chuyển đến tận cùng bảng gel (1-2 giờ). Nếu chạy gel có gradient muối sẽ cho phép đọc được nhiều base hơn trên một gel do việc làm ngắn lại khoảng cách giữa các băng ADN ở phần cuối bản gel. Nên dừng điện di từ 45 phút – 1 giờ. Tắt nguồn điện, bổ sung 1/2 thể tích 3M NaOAc vào bể đệm đáy, trộn đều. Lại tiếp tục công việc chạy gel cho đến khi BPB di chuyển đến cuối bản gel. Chú ý sự có mặt của muối nồng độ cao sẽ dẫn đến nhiệt độ tăng, do đó cần phải kiểm soát nhiệt độ của quá trình chạy gel nhằm tránh vỡ kính và hỏng gel. Chuẩn bị gel cho phát hiện bằng ảnh phóng xạ. 17, Lấy bản gel ra khỏi thiết bị, chú ý bể đệm đáy chứa chất phóng xạ, các nucleotid dư sẽ đi ra khỏi bản gel. 18, Đổ bỏ bể đệm trên. 19, Đặt bản gel trên bàn thí nghiệm sao cho tấm kính ngắn lên trên. Phủ lên một lớp SaranWrap, dùng đá lạnh đặt lên trên khoảng 2 phút để cho gel co lại, dùng thanh nhựa tách các lớp kính ra, giữ nguyên không cho tấm kính trên tiếp xúc lại với gel vì như vậy gel sẽ dính và bị vỡ. Lúc này gel sẽ chỉ dính vào tấm kính dưới không có silicon được phủ khi đổ gel. 20, Đặt tấm giấy Whatman lên trên gel và vuốt đều nhẹ nhàng, lúc đó gel sẽ dính vào giấy và có thể lấy ra dễ dàng (Chú ý trong trường hợp sử dụng chất phóng xạ là S35 thì nên cố định mẫu theo cách dưới đây). 21, Sau khi lấy gel ra hỏi tấm kính đáy thì bọc gel với SaranWrap, chú ý tránh không khí đi vào giữa lớp SaranWrap và gel, điều này sẽ cản trở ảnh phóng xạ trên phim. 22, Có thể đưa vào thiết bị sấy gel. 23, Sấy gel trong khoảng 45 phút tại 80oC. 24, Đặt phim X-ray theo đúng chỉ dẫn vào gel trong hộp cassette (có phim chỉ có một mặt thì mọi thao tác phải đúng để có ảnh phóng xạ). Thời gian nhận được bức xạ là 12-14 giờ đối với P32 và 24-48 giờ đối với S35. 25, Đọc kết quả. Cố định gel: Mục đích của bước này là loại bớt urea khỏi gel vì sự có mặt của urea sẽ hạn chế sự phân cách giữa các ADN, các bước được thực hiện như sau: + Đặt bản kính có gel vào khay chìm trong hỗn dịch 15% ethanol và 10% acetic acid. + Cố định trong 10-15 phút đủ để urea khuyếch tán khỏi gel. + Dùng giấy lọc để thấm hết dịch cố định khỏi gel. + Tiếp tục làm theo các bước như quy trình hiện ảnh phóng xạ. Hóa chất và cách chuẩn bị:Phương pháp: Giải trình tự ADN sợi kép sử dụng KIT biến tính nhanh với sequenase. Phương pháp này thích ứng với việc giải trình tự các plasmid sợi đôi. Bắt đầu bằng việc gây biến tính trong kiềm (NaOH) hoặc glycerol, sau đó kết tủa ADN và bắt cặp với ADN mồi dùng để giải trình tự. Phương pháp giải trình tự ADN với sợi đôi cần lượng ADN mồi cao gấp 10-20 lần so với sợi đơn. Việc giải trình tự gồm hai phản ứng: phản ứng đánh dấu và phản ứng dừng tại điểm cuối. Trong phản ứng đánh dấu các sợi ADN bổ sung được gắn kết với chất đánh dấu phóng xạ trong việc kéo dài chuỗi. Sau đó phản ứng kéo dài chuỗi ADN bị dừng một cách ngẫu nhiên do nồng độ thấp của nucleotid trong dịch phản ứng (0.08 mM). Bước tiếp theo là kết thúc chỗi với dideoxynucleotid khi có nồng độ cao của deoxyribonucleotid (33,3 mM). Các trình tự tiếp theo của sợi khuôn sẽ được xác định với nồng độ cao của deoxynucleotid trong khi phản ứng đánh dấu xảy ra trước việc bổ sung dideoxynucleotid vào hỗn hợp phản ứng. Quy trình sau đây đã được rút gọn, có nhiều phần tiện ích chi tiết hơn và thay đổi trong các tài liệu hướng dẫn. Điều quan trọng để có kết quả tốt trong việc giải trình tự ADN là chất lượng cao của ADN plasmid. Trong các khâu chuẩn bị việc xuất hiện các phân tử đứt gãy sẽ dẫn đến kết quả không rõ, khó xác định các băng. Các plasmid sợi đơn phải được làm biến tính cho mở xoắn trước khi giải trình tự. Biến tính các plasmid xoắn do liên kết hóa trị không phải lúc nào cũng đạt được bằng xử lý nhiệt tại nhiệt độ bắt cặp mồi hay trong đệm phản ứng, bởi lẽ nhiệt độ mở xoắn thường là 105ng là 105oC. Có hai phương pháp khá nhanh và tiện lợi: Một phương pháp xuất phát từ thực tế là glycerol hoặc ethylen glycerol có tác dụng làm giảm nhiệt độ mở xoắn, còn phương pháp thứ 2 là mở xoắn bằng môi trường kiềm. Cả hai phương pháp đều thực hiện đơn giản cho hầu hết các loại ADN khuôn. Đối với hai phương pháp biến tính ADN, nên dùng lượng ADN khuôn ở nồng độ 0.5 pmol (05- 0.3 mg) và lượng primer 2-5 pmpol. Nếu sử dụng thể tích ADN nhỏ hơn thì phải pha đến nồng độ thích hợp bằng nước. Kết quả tốt nhất thu được là theo tỷ lệ phân tử mồi : sợi khuôn là 4 : 1. Tỷ lệ này nên được duy trì khi tiến hành với lượng ADN khuôn khác nhau. Trong mọi trường hợp khi mà số lượng thành phần nào quá ít thì đều dẫn đến băng không rõ ở cuối bản gel. Lượng ADN khuôn cần theo phương pháp này ít hơn so với sử dụng sequenase KIT 2.0 do không bị mất ADN trong bước kết tủa với ethanol. Tác nhân gây biến tính ADN khuôn trong KIT này là đệm 40:50 glycerol và ethylen glycerol. Việc bổ sung đệm này vào dung dịch ADN theo tỷ lệ chỉ dẫn; 40% glycerol sẽ làm giảm nhiệt độ biến tính ADN xuống khoảng 20oC. Chý ý khi dùng đệm glycerol sẽ gây nên việc di chuyển không đều trên gel điện di tại vùng có khoảng cách 300-400 kể từ đoạn ADN mồi. Nên sử dụng đệm chạy gel hạn chế tác dụng của glycerol (thay taurin cho đệm chạy gel TBE) sẽ khắc phục được điều này. ADN plasmid sẽ biến tính tại bất kỳ nhiệt độ nào ở pH kiềm (pH 13). Điều quan trọng đối với phương pháp này là việc trung hòa bước biến tính kiềm bằng HCL phải được thực hiện một cách chính xác. Như vậy nên dùng một loại pipet cho việc bổ sung kiềm và trung hòa bằng acid HCL sau đó. Nói chung dung dịch NaOH và HCL được chuẩn bị có nồng độ 1M, trong trường hợp dùng hóa chất riêng phải đảm bảo nồng độ chỉ dao động trong khoảng 0,95- 1.05M. Bước gây biến tính kiềm và kết tủa với ethanol cũng có thể được thực hiện với các hóa chất trong KIT nhưng đôi khi cũng không đạt được kết quả mong muốn và cần phải cải tiến. Trong trường hợp giải trình tự với ADN sợi đơn, không phải thực hiện bước biến tính ADN, hỗn dịch được chuẩn bị đơn giản gồm: ADN plasmid, 05 pmol primer, 2 ml đệm phản ứng và bổ sung nước cho đủ 15 ml. Phản ứng tốt sẽ đọc được trình tự dài khoảng 400 bps. Cách tiến hành cụ thể:1. Biến tính ADN (theo hai cách trình bày ở trên).A. Biến tính với glycerol:ADN: 0.5 – 0.3 mg (thể tích tối đa 7 ml). Đệm biến tính plasmid: 5 ml. Primer: 1 ml. Nước cất dẫn đến: 13 ml. Trộn đều, ủ trong 5 phút ở 90 – 100 oC, làm lạnh nhanh trên đá. Bổ sung thêm đệm phản ứng: 2 ml để đạt được tổng thể tích là 15 ml. B. Biến tính với kiềm (không cần kết tủa với ethanol). Thành phần phản ứng gồm: ADN: 0.5 - 3mg (thể tích tối đa là 8ml) NaOH 1M: 2 ml. Primer: 1 ml. Nước cất dẫn đến: 11 ml. Trộn đều, giữ trong 10 phút tại 37oC, làm lạnh nhanh trên đá. Điều quan trọng trong phương pháp này là bước trung hòa phải được thực hiện chính xác với HCL. Sau đó bổ sung HCL 1M : 2 ml. Đệm phản ứng plasmid: 2 ml. Tổng thể tích là: 15 ml. 2. Bắt cặp mồi vào sợi khuôn: Giữ hỗn dịch ADN và mồi ở 37oC trong 10 phút, sau đó giữ trên đá lạnh (mẫu chỉ nên dùng trong vòng 4 giờ). 3. Chuẩn bị tube chứa hỗn dịch kết thúc chuỗi: Trong khi thực hiện bước bắt cặp sợi khuôn và sợi mồi, tiến thành hút 2,5 ml hỗn dịch kết thúc chuỗi cho từng loại (ddA, ddC, ddG, ddT) cho vào từng tube riêng biệt. Đậy nắp, để trên đá để dùng cho các bước 5 và 7. 4. Pha loãng dịch đánh dấu: Pha loãng dịch đánh dấu 5 lần với nước và giữ trên đá. Không nên pha loãng dịch đánh dấu khi muốn đọc đoạn ở xa mồi. Dịch đánh dấu: 2 ml Nước: 8 ml Tổng số: 10 ml. 5. Làm ấm các mẫu đá chuẩn bị trong bước 3: Bốn mẫu được chuẩn bị trong bước 3 lần lượt là ACGT được ủ ở 37oC trong 1 phút. 6. Phản ứng đánh dấu (kể từ bước này thì mọi đầu côn và pipét sẽ thực hiện với hóa chất phóng xạ, do đó cần phải có các biện pháp bảo vệ và đeo găng tay). Bổ sung lần lượt các thành phần sau vào 15 ml hỗn dịch bắt cặp đã được chuẩn bị trong bước 1. DTT 0.1M : 1 ml Dịch đánh dấu (bước 4): 2 ml Hóa chất phóng xạ: S35 (P33, P32): 0.5 ml Enzym T7 sequenase cho plasmid: 2 ml Tổng thể tích : 20.5 ml (Thể tích này sẽ được chia ra 4 phần trong bước 7) Trộn đều (tránh bọt), ủ hỗn dịch tại nhiệt độ phòng 2 -5 phút (đối với plasmid làm biến tính trong glycerol thì ủ 5-10 phút tại nhiệt độ phòng hoặc 5 phút tại 37oC). Thường bổ sung hỗn dịch T7 sequenase cho plasmid cuối cùng, sau khi các thành phần khác đã được bổ sung. Không bao giờ bổ sung hỗn dịch T7 sequenase cho plasmid vào hỗn dịch đánh dấu, DTT hay hỗn dịch khác không phải là đệm. Chú ý: Nếu trong trường hợp chất phóng xạ S35 để quá lâu thì có thể tăng lượng lên 2 ml cho mỗi phản ứng, sau đó giảm lượng nước tương ứng. 7. Phản ứng kết thúc chuỗi: a. Lấy 4,5 ml hỗn dịch thu được từ bước 6 cho vào thành trên của 4 tube kết thúc chuỗi đã được làm ấm ở bước 5 (A, C, G, T). Đậy nắp lại và li tâm nhanh để trộn đều, tiếp tục ủ trong 5 phút tại 37oC (có thể kéo dài tới 30 phút). Nếu thấy cần thiết thì có thể li tâm nhanh để thu lấy hỗn dịch trước khi tiến hành phản ứng đánh dấu. b. Tại thời điểm cuối của 5 phút, bổ sung 4 ml hỗn dịch dừng phản ứng vào thành trên của các tube và đóng nắp nhanh. Như vậy có thể dừng phản ứng tại thời gian thích hợp bằng cách li tâm để dịch dừng phản ứng đi xuống hỗn dịch phản ứng. c. Ngay lập tức li tâm để dịch dừng phản ứng đi xuống hỗn dịch phản ứng, sau bước này phản ứng đánh dấu đã hoàn tất và mẫu có thể được giữ ở 4 oC để sử dụng trong ngày hôm sau. d. Xử lý ở nhiệt độ 75oC cho các mẫu khoảng 2-3 phút ngay trước khi tra mẫu lên gel, với các mẫu chứa glycerol thì việc lên mẫu dễ dàng với đầu côn vàng. e. Lên khoảng 2-3 ml mẫu cho mỗi giếng trên gel giải trình tự. Nên đưa một lượng đồng mẫu trên các giếng. Cách tốt nhất để lên mẫu không bị bọt là hút 3 ml mẫu nhưng khi tra chỉ dùng 2 ml là đủ. Hóa chất:
Hỗn dịch kết thúc chuỗi:
Dịch dừng phản ứng: 95% Formamide 20mM EDTA (Na2), pH 8.0 0.05% Bromopheno Blue (BPB). 0.5% Xylen CyanolFF (XC). Chú ý, nếu bạn gặp trục trặc với việc giải trình tự ADN thì các plasmid nên được tái tinh sạch với Genee-Clean hoặc sắc ký trao đổi ion.
Hình 1.11. Tra mẫu trên thiết bị chạy gel. Các phần dưới đây đề cập đến một số vấn đề nảy sinh và cách giải quyết khi tiến hành giải trình tự ADN. - Muốn đọc được trình tự ADN dài hơn: Yêu cầu đầu tiên là phải đọc được đoạn gene đến mức có thể trên gel. Thông thường thì kích thước đọc cho mỗi phản ứng trên gel là 250-300 nucleotid. Với thời gian chạy gel lâu khoảng 8 giờ thì có thể đọc được kích thước gene dài hơn 400 nucleotid. Nếu sử dụng gel gradient sẽ không phù hợp cho mục đích này vì khoảng cách giữa các băng ADN sẽ hẹp tới mức không thể đọc được. Nồng độ gel acrylamid dao động 4-6% là thích hợp cho kết quả tốt. Có một giải pháp nữa là có thể sử dụng gel với gradient muối nhưng vẫn giữ nguyên thời gian chạy gel (xem phương pháp 2, bước 16). - Sự dồn các băng: Nguyên nhân chung để không thể hoàn tất việc giải trình tự ADN là các băng không rõ do cấu trúc bậc hai trong sản phẩm của phản ứng kéo dài chuỗi, điều này thường được mô tả là sự dồn ép các băng do các băng bị thu hẹp một cách bất thường trên ảnh fim của kết quả giải trình tự ADN. Các khoảng cách hoàn toàn như nhau khi mà khoảng 3 liên kết G-C bổ sung kèm với 2-5 base có thể dẫn đến kết quả không thể đọc được đoạn gene chiếm một khoảng nhỏ trên bản gel. Khoảng trống bất thường là do ảnh hưởng của chuỗi với khoảng cách ngắn của cấu trúc bậc 2 sẽ có cùng tốc độ di chuyển như là sợi thẳng có chiều dài ngắn, tại ví trí bản fim ngoài vùng dồn băng thì khoảng cách các băng sẽ rộng khác thường do sự kém bền của cấu trúc bậc 2 làm cho chiều dài của chuỗi tăng lên và sự di động của chuỗi trở nên như bình thường. Việc giải trình tự qua vùng gene dồn nén có thể được khắc phục bằng cách thực hiện việc đọc trình tự trên cả hai sợi. Sự bất thường về việc di chuyển nảy sinh ở các chuỗi có chiều dài không đủ cho việc bắt cặp. Điều này có nghĩa vùng gene có các băng không rõ ràng sẽ được khắc phục bằng cách sử dụng dITP thay cho dGTP trong phản ứng giải trình tự ADN. Lý do là base I chỉ có 2 liên kết với C thay vì 3 liên kết trong trường hợp với G, kết quả là ADN sẽ dễ duỗi hơn. Kỹ thuật này thường được dùng, do đó dITP có trong KIT sequenase. Một cách khác có thể làm kém bền vùng base dồn nén do còn cặp đôi đó là thực hiện chạy gel trong điều kiện biến tính (ví dụ sử dụng formamide trong nước dùng đổ gel). Trong trường hợp này khi thay thế 25% formamide cho nước sẽ đạt được kết quả tốt nhất. Gel có formamide thường được dùng khi vùng dồn nén khá gần mồi, nên xử lý formamide qua gel trao đổi ion ngay trước khi chuẩn bị gel. Trong các trường hợp mà các cấu trúc cặp đôi phức tạp hơn khi mà vùng dồn nén lại nằm ngay tại vị trí tương đồng trên sợi đối diện. Khi đó cách giải quyết sẽ trở nên khó khăn vì với các kỹ thuật hiện có thường đưa lại các kết quả khác nhau. - Cường độ các băng thiếu sự đồng nhất: Một vấn đề khác trong phản ứng giải trình tự ADN là cường độ các băng không đồng nhất. Điều này thường xuất hiện trong phương pháp giải trình tự với thiết bị tự động dựa vào phương pháp phát hiện bức xạ huỳnh quang. Sự thiếu đồng nhất là do sự có mặt của sequenase cần Mg++ gắn dideoxy kết thúc chuỗi khác nhau tại các vị trí khác nhau (vấn đề này được bàn luận kỹ trong tài liệu USB sequenase).
b. Kỹ thuật giải trình tự ADN trên thiết bị giải trình tự ADN tự động:
Hình 1.12. Thiết bị giải trình tự ADN tự độngg (3100-Avant Genetic Analyzer)
Trong phần này chúng tôi đề cập phản ứng giải trình tự ADN trên 2 loại thiết bị chủ yếu hiện đang được dùng phổ biến là: Beckman Coulter và ABI 3100-Avant Chuẩn bị mẫu giải trình tự ADN:1. Thành phần phản ứng (cho 1 phản ứng 20 ml):- Terminator Ready Reaction Mix*: 8 μl - Template: 80-100 ng ( với plasmid là: 300ng) - Primer : 3.2 pmol - Nước cất vô trình: đủ 20 μl Chuẩn bị Teminator Ready Reaction Mix cho 4 phản ứng: 15 μl Bigdye Sequencing Buffer 3.1 (5X) + 15 μl nước = 30 µl (2.5X) 20 μl đệm 2.5X + 20 μl Ready Reaction Premix= 40 μl Terminator Ready Reaction Mix (dùng 8 μl) cho một phản ứng. 2. Tiến hành phản ứng:Phản ứng được thực hiện trong 200 ml tube hay trong microplate. Cycling được thực hiện trong máy PCR theo chương trình sau: Denature 96oC: 1 phút 96 oC: 10 giây 50oC: 5 giây 60oC: 4 phút Cycling: 25 cycles. 3. Kết tủa sản phẩm PCR:- Cho vào 5µl EDTA 125 mM + 60µl E-OH 100%E-OH 100% - Trộn đều, để 15 phút ở nhiệt độ phòng. Ly tâm 15 000 v/phút - Rửa lại bằng 60µl E-OH 70% và lại ly tâm (4oC) - Làm khô mẫu ở nhiệt độ phòng (Speed Vac). - Khi mẫu khô, thêm 10µl Hi-Di formamide, trộn đều sau đó ủ ở 96oC trong 2 phút. - Để trên đá trước khi cho vào seq. 4. Đưa mẫu vào máy đọc trình tự.
Phòng thí nghiệm "ảo" : http://web.mit.edu/7.02/virtual_lab/PBC/PBC5virtuallab.html Formazan gồm MTT (3-(4, 5-dimethyl-2-thiazolyl)-2, 5-dihphenyl-2H-tetrazolium bromide) và nước hòa tan XTT (23-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide). |



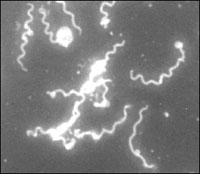














© http://vietsciences.free.fr và http://vietsciences.org Dương Văn Hợp và Nguyễn Lân Dũng